All published articles of this journal are available on ScienceDirect.

Pulmonary Arterial Pressure Response During Exercise in COPD: A Correlation with C-Reactive Protein (hsCRP)
Authors Info & Affiliations
Abstract
Background:
The non-invasive assessment of pulmonary haemodynamics during exercise provides complementary data for the evaluation of exercise tolerance in patients with COPD.
Methods:
Exercise echocardiography in the semi-supine position was performed in 27 patients with COPD (C) with a forced expiratory volume in one second (FEV1) of 36±12% predicted and 13 age and gender-matched non-COPD subjects (NC). COPD patients also underwent cardiopulmonary exercise testing with gas exchange detection (CPET). Furthermore, serum high sensitive C-reactive protein (hsCRP), a marker of systemic inflammation, was also measured.
Results:
The maximal work rate (WRmax) and aerobic capacity (VO2peak) were significantly reduced (WRmax: 77±33 Watt, VO2peak: 50±14 %pred) in COPD. Pulmonary arterial systolic pressure (PAPs) was higher in COPD versus controls both at rest (39±5 vs. 31±2 mmHg, p<0.001), and at peak exercise (72±12 vs. 52±8 mmHg, p<0.001). In 19 (70%) COPD patients, the increase in PAPs was above 22 mmHg. The change in pressure (dPAPs) correlated with hsCRP (r2=0.53, p<0.0001) and forced vital capacity (FVC) (r2=0.18, p<0.001).
Conclusion:
PAPs at rest and during exercise were significantly higher in COPD patients and correlated with higher hsCRP. This may indicate a role for systemic inflammation and hyperinflation in the pulmonary vasculature in COPD.
The study was registered at ClinicalTrials.gov webpage with NCT00949195 registration number.
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) seems to be not only a functional disorder of the lungs, but it may have systemic effects as well [1]. Extrapulmonary manifestations have been described in skeletal muscles, bones and the cardiovascular system [2]. It is postulated that low intensity chronic inflammation may result in oxidative stress and endothelial dysfunction, ischemic heart disease or stroke [3, 4].
The level of high sensitive C-reactive protein (hsCRP) may be a marker for systemic inflammation [5]. Higher value of hsCRP has been identified in chronic heart failure and cardiovascular disease [6]. Pulmonary hypertension (PH), a common finding in COPD, has a major impact on the quality of life and survival [7-9]. Arterial PH was associated with elevated serum level of CRP, IL-6, TNF and correlation was observed between C-reactive protein and endothelin-1 levels, showing the potential role of systemic inflammation in the pathogenesis of pulmonary hypertension in COPD [8, 10, 11].
The pathogenesis of PH in COPD is complex. Alveolar hypoxia results in pulmonary arterial vasoconstriction in order to provide optimal alveolar ventilation-perfusion ratio [7, 12]. Hypoxia may have both a direct and indirect effect on pulmonary vasculature (through transmitters and biogen amines) [8, 12]. Pulmonary hypertension is present in a substantial part of COPD patients (and is found in up to 50% according to diagnostic modalities). Otherwise, severe pulmonary hypertension is manifested only in 5% of these patients [13-16]. However, pulmonary pressure can increase significantly during exercise [17-19], which may have consequences in assessment of exercise capacity. Pulmonary pressure increment in an age-dependent way is presented in healthy subjects, otherwise higher increment in COPD may result higher load on circulation, heart and reduction of exercise tolerance.
We wished to determine whether the increment in PAPs during exercise was related to systemic inflammation in COPD as assessed by monitoring high sensitive CRP. Furthermore, we sought to clarify the correlation between PAPs increment during exercise and exercise physiologic variables. Since there is limited data in the literature on pulmonary arterial pressure response during exercise in healthy subjects, in this study, the change in pulmonary arterial peak systolic pressure (PAPs) during exercise in an age and gender-matched non-COPD subject group was also investigated.
MATERIAL AND METHODS
Study Subjects
Twenty-seven stable patients with COPD according to the GOLD criteria and a control group of thirteen age and gender-matched non-COPD subjects participated in the study (Table 1). The severity of airway obstruction ranged from severe to very severe in the COPD group [20]. COPD therapy with maximal bronchodilator effect was based on lung function related GOLD stages. Subjects were screened for hypertension, smoking history, hyperlipidemia and diabetes mellitus. Control subjects as an age-matched population had representative co-morbidities. Significant airway obstruction or reduction in diffusion capacity was not detected in the non-COPD group. 38 patients with COPD were initially screened, however eleven were excluded because the semi-supine echocardiography test could not be performed, and the tricuspid jet could not be visualized at peak exercise (Fig. 1). The study was approved by the Ethical Committee of University of Szeged and the patients gave written consent to their participation.
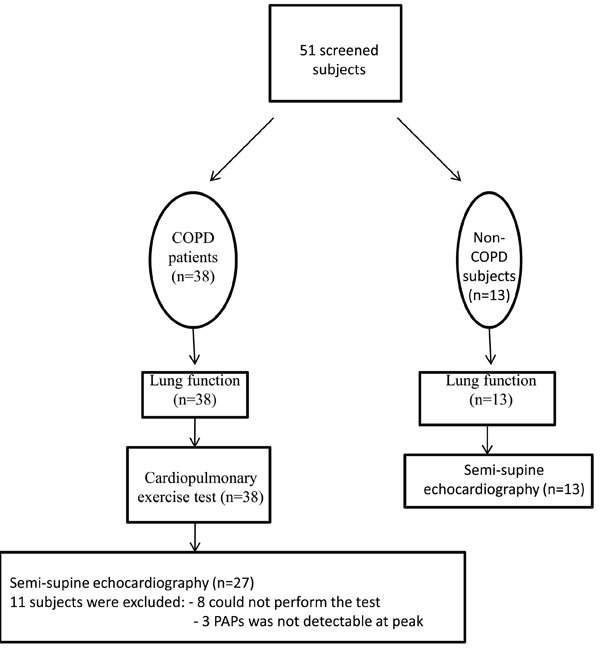
Flow of participants through each study stage.
Pulmonary Function and Exercise Testing
Patients performed a series of pulmonary function tests (PFT) (Vmax 229 and Autobox 6200, Sensormedics, Yorba Linda, California) including spirometry, body plethysmography and diffusion capacity measurement [20, 21]. Normal values were those of NHANES III standards [21]. Patients inhaled 400μg of salbutamol via a spacer 20 minutes before testing.
Demographics in patients with COPD and in healthy subjects (n=40).
| COPD patients n=27 | Non-COPD subjects n=13 | p-value | |
|---|---|---|---|
| Age (years), mean±SD | 58±10 | 63±5 | ns |
| Male sex(n) | 15 (56%) | 6 (46%) | ns |
| Height (cm), mean±SD | 167±10 | 170±10 | ns |
| BMI (kg/m2), mean±SD | 24±5 | 26±3 | ns |
| Diabetes n (%) |
5 (19) | 1 (8) | <0.001 |
| Hypertension n (%) |
14 (52) | 5 (38) | <0.05 |
| Smoking history (pack-year), mean±SD | 33±12 | 12±8 | <0.05 |
| Hyperlipidemia n (%) | 5 (19) | 2 (15) | ns |
Mean±SD, BMI=body mass index; SD=standard deviation
Immediately following the completion of PFTs, an incremental exercise test was performed on an electronically braked cycle ergometer (Ergoline-900, Marquette). After a 3-minute rest and a 3-minute constant pedalling at 20W, the work rate was increased 5 or 10 W/min in ramp profile. The ramp slope was: FEV1 <1.0Litre – 5W/min, FEV1 >1.0Litre – 10W/min. Pedalling rate was kept constant at approximately 60rpm. Pulmonary ventilation (‘VE), oxygen uptake (‘VO2) and carbon-dioxide output (‘VCO2) were measured breath-by-breath by a mass flow-sensor and exercise metabolic measurement system (Vmax29c, SensorMedics). Lactic acidosis threshold (LAT) was identified by the modified V-slope method [22]. Heart rate, 12-lead ECG (Cardiosoft, SensorMedics) and oxygen saturation by a pulsoximeter (SatTrak, SensorMedics) was monitored. Maximal voluntary ventilation was estimated as 40xFEV1 [23]. Breathlessness and leg fatigue were evaluated at peak exercise by modified Borg scale [24].
Resting and stress Doppler echocardiographic studies
Echocardiographic studies were performed with a commercially available echocardiographic system (Vivid 7 Expert, General Electric Healthcare). Resting cardiac dimensions were obtained by parasternal long-axis M-mode echocardiography, according to the American Society of Echocardiography standards [25]. These dimensions included left ventricular end-diastolic diameter, interventricular septal wall and posterior wall thickness, right ventricular outflow diastolic diameter, and left ventricular end-systolic diameter. Using the apical 4-chamber view, the M-mode cursor was placed through the junction of the tricuspid valve plane and right ventricular free wall. Tricuspid annular plane systolic excursion (TAPSE) was determined by the difference in the displacement of the right ventricular base during systole and diastole. Each parameter was measured off-line in triplicate and averaged. All images were obtained during held mid-expiration.
All patients and controls had normal left ventricular ejection fraction (EF > 55%) and no echocardiographic evidence of significant right ventricular dysfunction. In an effort to diagnose left ventricular diastolic dysfunction related heart failure, and secondary pulmonary PH, the E/A was recorded. Patients and the control subjects underwent a standardized multistage semi-supine bicycle exercise study in the recumbent position. Patients and controls pedalled at constant speed beginning at a workload of 25 W, with an increment of 25 W every 3 minutes. The protocol included both a low intensity "warm up" and a "cool-down" phase. Doppler evaluation of the tricuspid valve was performed in the apical four-chamber view. The tricuspid regurgitant jet was localized in the color mode, and then measured in the continuous wave mode at a sweep speed of 75 mm/sec. To ensure accurate transducer position throughout the exercise, the location of the continuous wave Doppler cursor was checked intermittently by two dimensional and colour imaging. We considered tricuspid insufficiency envelopes to be technically adequate when signals were pansystolic and showed well-defined borders. The maximum tricuspid insufficiency velocity was assigned to the highest coherent boundary on the spectral wave form. Maximum velocities recorded at rest, at each stage of exercise permitted the calculation of the transtricuspid gradient and PAPs. By a modification of the Bernoulli formula, the maximum transtricuspid gradient (in mmHg) was estimated as the product of 4 and the square of the maximum tricuspid insufficiency velocity [26]. PAPs was computed as the sum of the transtricuspid gradient and right atrial pressure (PAPs = 4V2+right atrial pressure) [27].
Lung function and resting and exercise echocardiography variables in patients with COPD and in non-COPD subjects (n=40).
| COPD patients (n=27) | Non-COPD subjects (n=13) | p-value | |
|---|---|---|---|
| Lung function variables | |||
| FEV1 (L) | 1.01±0.38 | 2.84±0.74 | <0.001 |
| FEV1 (%pred) | 36±12 | 103±14 | <0.001 |
| FVC (L) | 2.45±0.71 | 3.77±0.94 | <0.001 |
| FVC (%pred) | 73±18 | 113±20 | <0.001 |
| FEV1/FVC (%) | 42±11 | 77±6 | <0.001 |
| TLC (%pred) | 117±21 | 104±13 | <0.05 |
| VC (%pred) | 77±18 | 114±18 | <0.001 |
| IC (L) | 1.64±0.48 | 2.57±0.43 | <0.001 |
| FRC (%pred) | 164±42 | 111±18 | <0.001 |
| RV (%pred) | 199±57 | 100±18 | <0.001 |
| RV/TLC (%) | 60±10 | 37±6 | <0.001 |
| DLCO (%pred) | 41±15 | 88±5 | <0.001 |
| Transthoracic echocardiographic variables | |||
| LA diameter (mm) | 36±3 | 37±2 | ns |
| % LVEF | 65±2 | 63±2 | ns |
| LVPW thickness (mm) | 10.5±0.6 | 10.8±0.6 | ns |
| LVS thickness (mm) | 10.6±0.6 | 10.8±0.6 | ns |
| LV EDD (mm) | 47±3 | 48±4 | ns |
| LV ESD (mm) | 27±4 | 27±3 | ns |
| TAPSE at rest (mm) | 26±5 | 22±4 | ns. |
| TAPSE at peak (mm) | 36±8 | 32±5 | ns. |
| dPAPs (mmHg) | 33±10 | 20±8 | p<0.001 |
Data are presented mean±SD; FEV1: forced expiratory volume in one second; FVC: forced vital capacity; TLC: total lung capacity; VC: vital capacity; IC: inspiratory capacity; FRC: functional residual capacity; RV: residual volume; DLCO: diffusion capacity of carbon monoxide; LA: left atrium; LVEF: left ventricular ejection fraction; LVPW: left ventricular posterior wall; LVS: left ventricular septum; LVEDD: left ventricular end-diastolic diameter; LVESD: left ventricular end-systolic diameter, TAPSE: tricusp annular plane systolic excursion; dPAPs: change of pulmonary arterial systolic pressure during exercise
Right atrial pressure was estimated at rest by the response of the inferior vena cava to deep inspiration and was assumed to be constant throughout exercise. With the trailing-edge to leading-edge technique, maximum inferior vena cava diameters before inspiration and minimum diameters after inspiration were measured in the subcostal view within 2 cm of the entrance to the right atrium. When the diameter of the inferior vena cava decreased by less than 50% after deep inspiration, the right atrial pressure was defined as 15 mmHg, and when the diameter decreased by more than 50%, the right atrial pressure was defined as 5 mmHg [28]. This technique, which assumes that the estimated RA pressure remains constant during exercise, has been validated against simultaneous measurements of pulmonary artery systolic pressure during incremental exercise [29]. TAPSE was determined and registered at peak exercise. Measurements were recorded with simultaneous electrocardiography. All data were stored digitally and the measurements were made off-line at the completion of each study. Echocardiographic studies were performed by an experienced cardiac sonographer, who had no knowledge of the clinical data.
Inflammation marker determination
Systemic inflammation marker high sensitive CRP was determined from serum with an immunoassay method [30].
Statistical analysis
Groups were compared by Student’s t-test and chi-square test in case of continuous and categorical variables, respectively. Significance was accepted at p<0.05. Distribution around the mean was expressed ± S.D. Distributions were tested for normality by Kolmogorov–Smirnov test and significance was accepted if p<0.05. Pearson correlations with dPAPs (change of systolic pulmonary arterial pressure during exercise) and dPAPs/WR (work rate) (change of systolic pulmonary arterial pressure during exercise per equivalent work unit) were also evaluated. An analysis of covariance (ANCOVA) was used to compare the dependence of some variables on the dPAPs in the two groups. Stepwise linear discrimination analysis was performed to examine the significance of the two groups based on several variables. In patients with COPD, two groups were separated based on systolic pulmonary arterial pressure increment. The cut-off point calculation as an abnormal pulmonary hemodynamic response (dPAPs>22mmHg) was based on systolic pulmonary arterial pressure increment during exercise using an equation of mean pulmonary pressure (mPAP=PAPs x 0.61+2mmHg) [31] and 15 mmHg mPAP elevation during exercise in >50 year-old healthy subjects was reported by Kovacs G et al. [32].
RESULTS
Demographics of the study subjects are presented in Table 1. Smoking history varied significantly between the two groups. COPD patients had several co-morbidities, and few subjects in the age-matched control group had hypertension, diabetes or hyperlipidemia (Table 1).
Lung function showed severe obstruction and hyperinflation at baseline in our subjects with COPD (Table 2). Resting transthoracic echocardiographic variables are presented in (Table 2).
Exercise physiologic variables showed reduced peak work rate (54±21%pred) and oxygen uptake (VO2:50±14%pred, VO2/kg:60±17%pred) with not high ventilatory equivalents (VE/VO2:36±5, VE/VCO2:39±7) in COPD. However, clinically important desaturation was not detected in COPD (Sat:93±2 vs. 91±4%).
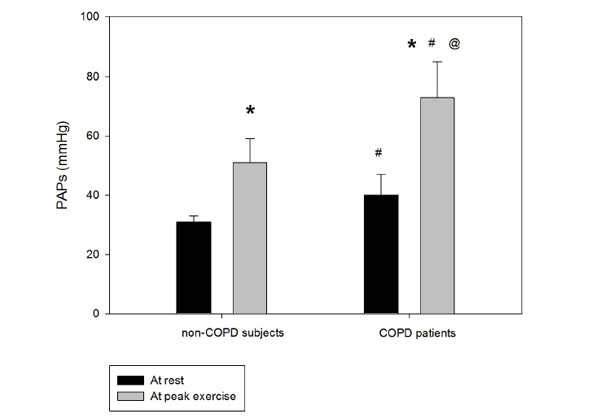
The increase of peak pulmonary arterial systolic pressure during exercise during semi-supine exercise echocardiography. PAPs: pulmonary artery systolic pressure; *:p<0.05 peak vs. rest, #:p<0.05 COPD patients vs. non-COPD subjects, @:p<0.05 peak-rest value difference between groups, error bars represents ±SE.
Systolic pulmonary artery pressure was significantly higher in the COPD group compared to the control subjects at rest, and the increase during exercise was significantly different between the groups, achieving higher value in the COPD group (Fig 2). The right ventricular function (TAPSE value) was normal at rest and remained physiologic at peak exercise in both groups (Table 2). The increment in the pulmonary arterial pressure during exercise correlated with the systemic inflammation marker hsCRP (Fig 3). In 19 COPD patients, the PAPs increment (dPAPs) was above 22 mmHg, and hsCRP was 11.1±8.8 mg/L compared to the 8 patients with PAPs increment (dPAPs) below 22 mmHg and lower hsCRP values (1.9±3.2 mg/L). The degree of obstruction (FEV1, FEV1/FVC) was not a predictor for the PAPs rise during exercise in the COPD group (Table 3). In the lung function variables, FVC as an indirect marker of hyperinflation correlated with dPAPs in the COPD group (Fig 4).
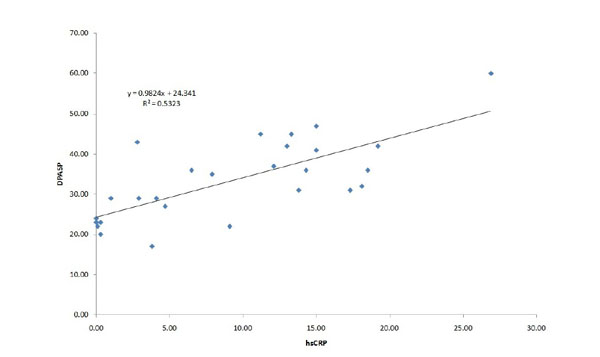
Correlation between increment of peak pulmonary artery systolic pressure during exercise and systemic inflammatory marker in patients with COPD. dPAPs: change in PAPs during exercise, hsCRP: high sensitive C-reactive protein, r2: linear regression correlation coefficient.
DISCUSSION
The pulmonary artery pressure response in a COPD and an age and a gender-matched control group was evaluated during exercise. The pulmonary artery pressure was significantly higher at rest and increased more in COPD compared to control age and gender-matched non-COPD subjects. The increase of pulmonary arterial pressure during exercise correlated with the systemic inflammatory marker hsCRP, and also with FVC in patient with COPD.
Secondary pulmonary hypertension in COPD results from sustained vasoconstriction and structural alterations to the pulmonary vascular bed. The major stimuli that are presumed responsible for these changes are chronic alveolar hypoxia, chronic inflammation and excessive shear stress [33]. Systemic or local inflammation may enhance the injury of acinar structure. Regardless of the stimuli that cause pulmonary hypertension, the structural changes that are thought to underlie the increased vascular resistance can be broadly classified into two processes: first, remodelling of the pulmonary resistance vessels and, second, a reduction in the total number of blood vessels in the lungs [33].
The serum CRP levels seem to correlate with the degree of systemic inflammation in COPD. A high level of CRP can also indicate an increased risk of cardiovascular diseases [34]. The systemic effect on the vasculature in COPD may also increase atherosclerosis induced stroke [35]. Indeed, the diastolic dysfunction related heart failure with E/A<1 and higher hsCRP might be partially responsible for the development of PH in our study as well.
In patients with COPD, the concomitant features of hypercholesterolemia, obesity, diabetes mellitus, hypertension, as a clinical feature of metabolic syndrome (a cause of systemic vascular disease) may also be accompanied by high CRP level [36]. In our investigation, many patients (about 50%) suffered from hypertension, and many had both hypertension and diabetes.
Pulmonary arterial pressure generally increases with age [32]. In healthy individuals, it is suggested that the normal mean pulmonary arterial pressure measured by right heart catheterization is 14.0 ± 3.3 mmHg at rest, and the age-dependent (especially > 50 years) increment during exercise may achieve a mean pressure of 30 mmHg [32]. Age-matched control subjects were involved in our investigation to exclude the age-dependent PH differences between the COPD and the non-COPD groups. In accordance with previous data [37]. PAPs increase during exercise by echocardiography was found in healthy subjects as well.
Pulmonary vascular impairment, secondary pulmonary hypertension may partially be responsible for the reduced oxygen uptake [38], exercise capacity and increased ventilatory equivalent for CO2. Analyzing gas exchange during cardio-pulmonary exercise test offers potential explanations for exercise limitation [39]. Reduced exercise tolerance with ventilatory inefficiency according to higher ventilatory equivalent for CO2 was detected in our COPD patients.
Pearson’s correlation between identified study variables and dPASP. dPASP/WR in the COPD group.
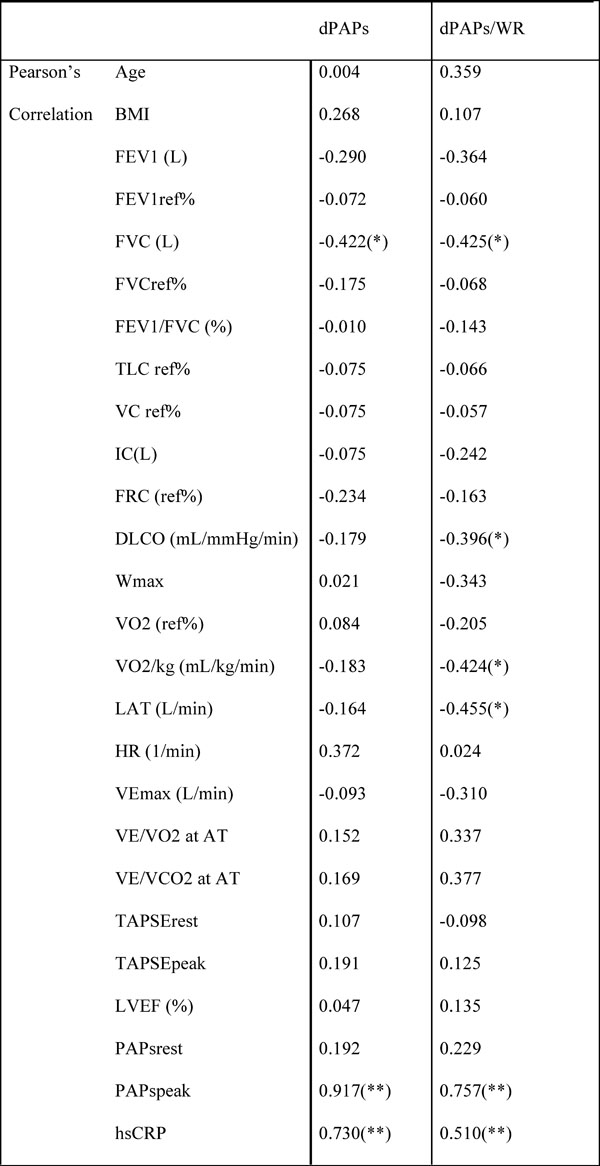 |
* Correlation is significant at the 0.05 level (2-tailed).
FEV1: forced expiratory volume in one second; FVC: forced vital capacity; TLC: total lung capacity; VC: vital capacity; IC: inspiratory capacity; FRC: functional residual capacity; RV: residual volume; DLCO: diffusion capacity of carbon monoxide; WR: work rate; VO2: oxygen uptake; VO2/kg: specific aerobic capacity; VCO2: carbon-dioxide output; LAT: lactate anaerobic threshold; VEmax: maximal minute ventilation; VE/VO2: ventilatory equivalent for O2; VE/VCO2: ventilatory equivalent for CO2; RER: respiratory exchange ratio; TAPSE: tricuspidal annular plane systolic excursion; LVEF: left ventricular ejection fraction; PAPs: pulmonary artery systolic pressure; hsCRP: high-sensitive C-reactive protein.
Exercise echocardiography in semi-supine position can be used to assess pulmonary arterial systolic pressure response to exercise which helps in better understanding the functional impairment in relation to dyspnoea during exertion. This type of non-invasive assessment of pulmonary hemodynamics is appropriate for early detection of pathologic changes in the pulmonary vasculature [40]. In our study, all pulmonary arterial pressure measurements were performed by only one experienced observer to eliminate the inter-observer variability.
Nevertheless, our study had limitations. Accurate measurement of pulmonary arterial pressure requires right heart catheterisation. The measurement of pulmonary arterial systolic pressure by echo-cardiography may be limited during exercise. 8 of 38 patients with COPD could not perform the semi-supine position exercise test, and PAPs was not detectable in 3 of them. The evaluation of PAPs value by echocardiography in special cases might be higher during exercise compared to direct PAPs measurement by right heart catheterisation [41]. Moreover, echocardiography results in false-positive detection of pulmonary hypertension in up to 50% of COPD patients [42]. Correlation analyses between hsCRP, FVC and dPAPs could show potential associations but by no means do reflect causality.
The theory of systemic inflammation might be only one component related to elevated pulmonary arterial pressure during exercise in our patients with COPD. PH manifested due to COPD and/or hypoxia: the pathobiological and pathophysiological mechanisms involved in this setting are multiple and include hypoxic vasoconstriction, mechanical stress of hyperinflated lungs, loss of capillaries, inflammation, and toxic effects of cigarette smoke. Moreover, there are data supporting an endothelium-derived vasoconstrictor-vasodilator imbalance [43]. As hypertension was quite frequent in both groups (52% and 38%), thereby left ventricular diastolic dysfunction might have contributed to PAPs elevation during exercise. Hyperinflation might have also during exercise been involved in the increase of PAPs as there was a negative correlation between numeric value of FVC and dPAPs.
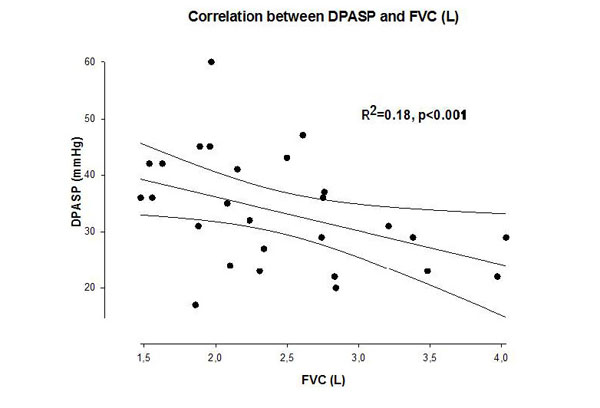
Correlation between increment of peak pulmonary artery systolic pressure during exercise and forced vital capacity in patients with COPD. dPAPs: change in PAPs during exercise, FVC: forced vital capacity, r2: linear regression correlation coefficient.
In summary, the pulmonary arterial pressure was higher at rest and increased significantly more during exercise in COPD patients compared to non-COPD control subjects. The rate of pulmonary arterial pressure increment partially correlated with the systemic inflammatory marker hsCRP. The increase in pulmonary arterial systolic pressure during exercise may in part be related to systemic inflammation. In case of high PA pressures during exercise, in order to maintain stroke volume the work of the right ventricle is disproportionally increased. Hemodynamic exercise testing with Doppler echocardiography may contribute to our better understanding of the exercise limitations in COPD and help define a more appropriate training program for the rehabilitation of patients with COPD.
LIST OF ABBREVIATIONS
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
This study was supported by funds available to the Department of Pulmonology, University of Szeged, Deszk, Hungary, and Department of Internal Medicine, Elisabeth Hospital, Hodmezovasarhely, Hungary.
The authors thank Prof. Sean Gaine, MD, PhD, Mater Misericordiae University Hospital, Dublin, Ireland for the meaningful suggestions and help to interpret the data of this study. The authors also thank Regina Palfoldi MD, Department of Pulmonology, University of Szeged to collect COPD patients for the study, Tunde Oravecz and Eva Asztalos to perform the lung function tests for the patients.

