All published articles of this journal are available on ScienceDirect.

Pathophysiology of Acute Respiratory Failure by CoV-2 Infection: Role of Oxidative Stress, Endothelial Dysfunction and Obesity
Authors Info & Affiliations
Abstract
Coronavirus disease 2019 (COVID-19) due to CoV-2 (coronavirus type 2) virus possess a particular risk of developing acute respiratory distress syndrome (ARDS) or SARS (severe acute respiratory syndrome coronavirus 2)-CoV2 in people with pre-existing conditions related to endothelial dysfunction and increased pro-inflammatory and pro-oxidant state. In between these conditions, chronic systemic inflammation related to obese patients is associated with the development of atherosclerosis, type 2 diabetes, and hypertension, comorbidities that adversely affect the clinical outcome in critical patients with COVID-19. Obesity affects up to 40% of the general population in the USA and more than 30% of the adult population in Chile. Until April 2021, 1,019,478 people have been infected, with 23,524 deaths. Given the coexistence of this worldwide obesity epidemic, COVID-19 negative outcomes are seriously enhanced in the current scenario. On the other hand, obesity is characterized by endothelial dysfunction observed in different vascular beds, an alteration which can be associated with impaired vasodilation, oxidative stress, and inflammatory events. Emerging evidence shows that obesity-related conditions such as endothelial dysfunction are associated with detrimental outcomes for COVID-19 evolution, especially if the patient derives to Intensive Care Units (ICU). This implies the need to understand the pathophysiology of the infection in the obese population, in order to propose therapeutic alternatives and public health policies, especially if the virus remains in the population. In this review, we summarize evidence about the pathogeny of Cov-2 infection in obese individuals and discuss how obesity-associated inflammatory and prooxidant status increase the severity of COVID-19.
1. INTRODUCTION
In Chile, the number of infected patients with Coronavirus disease 2019 (COVID-19) reached 1.000.000 in April 2021 with more than 23.000 deaths, threatening the health system collapse due to the increasing demand for ICU. Emerging evidence suggests that obesity related conditions seem to worsen the effect of the virus respiratory infection, especially at ICU admission. Studies from Chinese cohorts of patients with COVID-19 have identified several risk factors for severe COVID-19, including age, hypertension, type 2 diabetes, and cardiomyopathy and ischemic heart disease [1]. However, obesity as a risk factor for detrimental outcomes in patients with COVID-19, has not been fully evaluated.
The experimental and clinical evidence indicates that obesity-induced endothelial dysfunction, with a simultaneousup-regulation of pro-inflammatory cytokines. Also, oxidative stress is a deleterious factor that systemically increases during obesity and promotes the development of diabetes, atherosclerosis, and endothelial dysfunction. In this view, some studies show higher levels of oxidative markers in obese patients with these risk factors [2, 3]. Based on this evidence, we propose that the endothelial dysfunction degree in obese patients with COVID-19 will determine, at the admission of ICU, a detrimental clinical evolution and poor prognosis. Therefore, the study of the pro-inflammatory profile and the level of oxidant stress in plasma of these patients during respiratory infection by SARS-CoV-2 are highly relevant for an assertive prognosis, therapy and eventual post-discharge management [4].
The systemic and molecular pathophysiological mechanisms that explain the interaction between obesity and severity due to SARS-CoV2 infection are varied; this review attempts to provide a limited vision regarding oxidative stress and inflammation markers.
1.1. Pathophysiology of Lung Injury Induced by SARS-CoV-2 Infection
Infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in humans is associated with a broad spectrum of clinical respiratory syndromes, ranging from mild upper airway symptoms to progressive life-threatening viral pneumonia [5]. Clinically, patients with severe COVID-19 have labored breathing and progressive hypoxemia and often need mechanical ventilatory support. Radiographically, peripheral lung
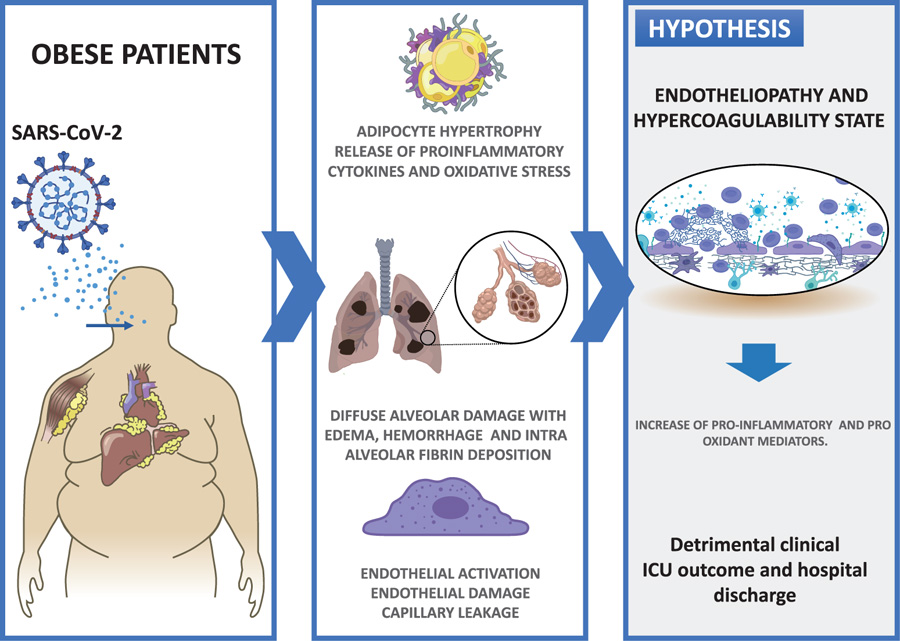
ground-glass opacities on computed tomographic (CT) chest imaging fulfill the Berlin criteria for ARDS [6]. Histologically, the hallmark of the early phase of ARDS is diffuse alveolar damage with edema, hemorrhage, and intra-alveolar fibrin deposition [7]. Diffuse alveolar damage is a nonspecific finding, since it may have non-infectious or infectious causes, including Middle East respiratory syndrome coronavirus (MERS-CoV), SARS-CoV, SARS-CoV-2 and influenza viruses [8]. Among the distinctive features of COVID-19 are the vascular changes associated with the severity disease. Clinically, the endothelial activation as expressed by elevated D-dimer levels, as well as cutaneous changes in the extremities, suggests thrombotic microangiopathy [9]. Disseminated intravascular coagulation (DIC) and large-vessel thrombosis have been linked to multisystem organ failure [10]. Peripheral pulmonary vascular changes are less well characterized; however, vasculopathy in the gas-exchange networks, depending on its effect on the ventilation/perfussion mismatching, could potentially contribute to hypoxemia.
Interestingly, the pathophysiology for COVID-19-related systemic micro-thrombosis, complicated by multiorgan failure, maybe specific and, in particular, different from DIC. Indeed, in contrast to sepsis-induced coagulopathy, consumption of platelets, coagulation factors and fibrinogen as well as bleeding complications are rare in severe COVID-19 patients, suggesting that DIC is not a common complication of the disease [11].
In summary, to establish explanatory bonds between the puzzling concepts of COVID-19, these disorders may be due to hypoxia combined with an immuno-triggered thrombo-inflammation supported by both an endotheliopathy and a hypercoagulability state. Taking into account that the activation of the endothelium would be a relevant step, markers such as oxidative stress could show early changes at the level of the pulmonary and the other organ microvasculature (Fig. 1).
1.2. Ventilatory Patterns in Obese Patients with SARS-CoV-2 Pneumonia
In SARS-CoV-2 pneumonia that required mechanical ventilation, two different phenotypes have been described [12]. The L phenotype (Low) presents with low elastance and high compliance, low ventilation/perfusion ratio, and decreased recruitability; in this case, the factor limiting pulmonary function will be perfusion. Moreover, the H phenotype (High) presents with high elastance and low compliance, pulmonary shunts, and high recruitability; the factor limiting pulmonary function will be ventilation [13].
The pathophysiological transition from Type L to Type H may be due to the evolution of COVID-19 pneumonia on the one hand and the injury attributable to high-stress ventilation in obese patients [14]. The possible key feature which determines the evolution of the disease, other than the severity of the disease itself, is the depth of the negative intrathoracic pressure associated with the increased tidal volume in spontaneous breathing. Indeed, the combination of a negative inspiratory intrathoracic pressure and increased lung permeability due to inflammation results in interstitial lung edema. This event, initially described by Mascheroni y cols., in an experimental setting, founding a fall in PaO2, a decrease in the static compliance of the respiratory system, and abnormal chest infiltrate [15]. These physical and molecular secondary events have been recently recognized as the leading cause of patient self-inflicted lung injury (P-SILI) [16]. When lung edema reaches a certain magnitude, the gas volume in the lung decreases, and the tidal volumes generated for a given inspiratory pressure decrease, leading to severe hypoxemia [17]. At this stage, dyspnea develops, which in turn leads to worsening P-SILI. In summary, if not available, signs which are implicit in Type L and Type H definition could be used as surrogates: respiratory system elastance and recruitability. Understanding the correct pathophysiology is crucial to establishing the basis for appropriate treatment.
In our intensive care unit, we have observed an unexplained increased prevalence of patients with obesity and SARS-CoV-2 pneumonia with ventilatory phenotype H (acute respiratory distress syndrome–like). From the ventilatory point of view, the patients need high positive end-expiratory pressure and respond well to recruitment maneuvers. This ventilatory pattern has been associated with a higher obesity in our clinical cohort and high cardiopulmonary comorbidities (hypertensive disease and chronic obstructive pulmonary disease) [18].
The incidence of ARDS is increased in patients with obesity [19]. To date, there are no studies that mention the association between the ventilatory phenotypes of COVID-19 and obesity. We suggest that understanding these relationships is crucial and clinically important in the treatment of patients with obesity and COVID-19 pulmonary complications.
1.3. Endothelial Dysfunction as a Link between Obesity and Severity of SARS-CoV-2 Infection
Obesity is a chronic inflammatory state associated with dysregulated endocrine and paracrine actions of adipocyte-derived factors, which in turn disrupt vascular homeostasis and cause endothelial dysfunction [20]. While the mechanisms by which obesity exacerbates COVID-19 infection are not fully understood, endothelial dysfunction may be the common link [21]. The endothelium is the cell layer lining the luminal surface of the blood vessels and, in healthy individuals, is a major regulator of vascular homeostasis. The main function of the endothelium is the barrier controlling molecules and cells transport from the bloodstream to the vessel wall and vice versa. The endothelium responds to a series of chemical and biomechanical cues by secreting factors regulating vascular tone, Smooth Muscle Cells (SMC) proliferation and migration, immune cell adhesion, thromboresistance, and vessel inflammation [22]. Cardiovascular risk factors such as hypertension and diabetes mellitus can activate the endothelium, resulting in the expression of chemokines, cytokines, such as interleukin (IL)-1, IL-6, IL-8, Monocyte Chemoattractant Protein-1 (MCP-1), and adhesion molecules (e.g. vascular adhesion molecule, VCAM-1; intracellular adhesion molecule, ICAM-1, E-selectin) that attract and facilitate immune cell extravasation [23]. Critical to the activation of Endothelial Cells (ECs) is the switch from Nitric Oxide (NO) signalling to Reactive Oxygen Species (ROS) signaling. Endothelial-derived NO promotes homeostasis and maintains the vascular wall in a quiescent state by the inhibition of proinflammatory cytokine secretion, immune cell extravasation, SMC proliferation and thrombosis, preventing vascular leakage. However, excessive ROS induces NF-κB signaling, the main regulator of inflammation [24]. This state of oxidative stress is a common underlying mechanism for endothelial cell dysfunction in response to biochemical and biomechanical pathophysiological stimuli [25]. Thus, endothelial dysfunction is considered one of the pathogenetic mechanisms of a whole range of inflammatory diseases [26, 27].
The pathogenetic role of the endothelial-derived factors evidences the impairment of their vital physiological properties. Thus, the detection of specific biochemical markers in the blood is an effective way for endothelial dysfunction diagnosis and characterize the vascular endothelium state. For example, in influenza virus infection, acute lung injury is associated with epithelial damage and impaired permeability in animal models and human serum markers of cardiac dysfunction [28]. However, the main clinical devastating effects are caused by endothelial dysfunction, thought to be the main mechanism leading cause for pulmonary oedema, respiratory failure and cardiovascular collapse [29]. In the case of SARS-CoV2 infected patients, the endothelial dysfunction would be caused by both direct virus cytopathic effect and inflammatory reaction, leading to a pro-thrombotic setting [29, 30]. Furthermore, the immune response-induced cytokine storm, the local and systemic inflammatory response responsible for an endotheliopathy and a hypercoagulability state, leading to both systemic and macro- and micro-thrombosis [31]. However, the exact pathophysiological mechanisms leading to severe pulmonary vascular dysfunction in obese patients and ARDS have not been elucidated.
1.4. Role of Inflammation in COVID-19
Inflammation plays a key role in the development of COVID-19 [32]. Sensors of viral infection and cellular damage (i.e. inflammasomes; TLR) trigger myeloid cell-dependent production of inflammatory cytokines (i.e. IL-1; IL-6; chemokines). Macrophages and inflammatory cytokines amplify local and systemic inflammation and are major drivers of organ failure [33]. Also, some experimental studies demonstrate the relationship between high levels of IL-6 and the degree of endothelial dysfunction [34, 35]. For example, some models of heart failure show an association of disease progression or organ damage (renal or myocardial fibrosis) with higher plasma levels of IL-6 and IL-10 [36, 37]. These support the clinical effects of antagonizing IL-6 pathway; in fact, blockade therapy using a humanized anti-IL-6 receptor antibody, tocilizumab, was observed in patients with cytokine release syndrome complicated by T-cell engaged therapy [38, 39]. Regarding SARS-CoV-2 disease, in REMAP-CAP and RECOVERY, the two largest clinical trials of IL-6 blockade and the only trials to show a mortality reduction, the benefit seemed predominantly in patients who received steroids. Hence, the contribution of negative studies that examined IL-6 blockade in the absence of routine administration of steroids is very useful but more challenging to interpret at this stage [40]. Lately, establishing whether there is a class effect or dose-effect of IL-6 blockade on improving mortality is a crucial question to answer. Indeed, both trials add important safety data to the literature, bolstering estimates of the relative short-term safety of these agents in diverse settings and diverse populations.
Understanding the relationship between COVID-19, obesity, pre-existing endothelial dysfunction, and observed endothelial injury would be imperative to develop new therapeutic targets to steer the trajectory of this pandemic. Indeed, anti-inflammatory therapies that block NF-κB and activate Nrf2 pathways, as well as novel therapies that address COVID-19 pneumonia and ARDS with disseminated intravascular coagulation, including anticoagulation, will be tested in obese patients [41]. In relation to this objective, it has been found that the maximal level of IL-6, followed by CRP level, was highly predictive of the need for mechanical ventilation. This suggests the possibility of using IL-6 or CRP level to guide escalation of treatment in patients with COVID-19-related hyperinflammatory syndrome [42].
1.5. Oxidative Stress and COVID 19
The association of Oxidative Stress (OS) markers with some viral or bacterial respiratory infection severity, such as respiratory syncytial virus, are well-established [43, 44]. Regarding the SARS-CoV-2 virus, the clinical data is finite, even though some lines of evidence propose that the ROS overproduction along with depletion of antioxidant functions have an important role in the pathogenesis of COVID-19 accompanied by severe respiratory failure [45]. ARDS animal models exhibited an increase in ROS levels with a reduced antioxidant capacity in the time of infection with SARS-CoV-2 [46, 47]. The lung injury severities of infected SARS-CoV-2 patients are speculated to be in association with some transcriptional redox factors, including NF-κB that are activated as a result of OS coupling with innate immunity, worsening the pro-inflammatory response of the host endothelial cells and induce lung injury [48, 49]. Some intracellular processes regulated by NF-kappaB account for alterations in mitochondrial bioenergetics that also increase intracellular ROS production and can induce cell death by apoptosis [50, 51]. Therefore, measurement of the activity of this factor could support the link of inflammation and oxidative stress with the severity of SARS-CoV2 infection.
Regarding some markers of OS, COVID-19 patients with moderate and severe symptoms showed elevated ROS levels accompanied by a decrease in total glutathione (GSH) and a higher ratio of oxidized/reduced glutathione; while the patients with milder symptoms maintain normal GSH levels [52]. Regarding extracellular antioxidants, the homeostasis between the mechanisms of the antioxidant defense and oxidative stress indicates the susceptibility ratio of the vascular cells to ROS [53, 54]. For example, depriving endogenous antioxidant protection in animals increases the lethality of coronavirus. At the same time, experimental animals with deleted components of ROS-generating machinery may be more resistant to respiratory viruses [48, 55]. However, to date, there are no clear analyses of plasma redox markers levels in COVID-19 patients. Moreover, during critical ill patients evolution, it is very important to determine if redox changes are in agreement with the time course of hypoxemic acute respiratory failure. In view of this evidence, the analysis of the clinical progression of the disease and related it with redox markers in critically ill obese COVID-19 patients is a new mechanistic paradigm of SARS CoV2 infection study. Regarding specific targets in oxidative stress and vascular involvement in COVID, our group recently commented on the protective effects at the vascular level, which could support some attenuation effect in the vasculopathy caused by SARS-CoV2 infection [56]. These effects would be based on antioxidant mechanisms triggered by melatonin.
CONCLUSION
Although the pathophysiology of obesity is known, oxidative stress and inflammation phenomena associated with the progression and severity of SARS-Cov2 infection could determine a therapeutic window in those patients who develop severe pneumonia and respiratory failure due to COVID -19 [57].
The obese patients express basal endothelial dysfunction. The time course of inflammation and oxidation markers with endothelial dysfunction could allow predicting obese patients susceptible to ventilatory complications and organ dysfunction. Thus, it will be relevant to evaluate the cardiopulmonary function and the association with biomarkers of oxidative stress, inflammation and endothelial dysfunction in order to optimize a prognostic score in obese patients.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

