All published articles of this journal are available on ScienceDirect.

New Concepts in the Invasive and Non Invasive Evaluation of Remodelling of the Right Ventricle and Pulmonary Vasculature in Pulmonary Arterial Hypertension
Authors Info & Affiliations
Abstract
Pulmonary arterial hypertension (PAH) is a rare fatal disease defined as a sustained elevation of pulmonary arterial pressure to more than 25 mmHg at rest, with a mean pulmonary-capillary wedge pressure and left ventricular enddiastolic pressure of less than 15 mmHg at rest. Histopathology of PAH is founded on structural modifications on the vascular wall of small pulmonary arteries characterized by thickening of all its layers. These changes, named as vascular remodelling, include vascular proliferation, fibrosis, and vessel obstruction. In clinical practice the diagnosis of PAH relies on measurements of pulmonary vascular pressure and cardiac output, and calculation of pulmonary vascular resistances. Direct evaluation of pulmonary vascular structure is not routinely performed in pulmonary hypertension since current imaging techniques are limited and since little is known about the relationship between structural changes and functional characteristics of the pulmonary vasculature. Intravascular ultrasound studies in patients with pulmonary hypertension have shown a thicker middle layer, increased wall-thickness ratio and diminished pulsatility than in control patients. Optical Coherence Tomography, a new high resolution imaging modality that has proven its superiority over intravascular ultrasound (IVUS) for the detection and characterization of coronary atherosclerotic plaque composition, may potentially be a useful technique for the in vivo study of the pulmonary arterial wall. In addition current progress in Echo Doppler technique will quantify right ventricular function with parameters independent of loading conditions and not requiring volumetric approximations of the complex geometry of the right ventricle. This would allow the in vivo study of right ventricular and pulmonary artery remodelling in PAH.
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a rare fatal disease defined as a sustained elevation of pulmonary arterial pressure to more than 25 mmHg at rest, with a mean pulmonary-capillary wedge pressure and left ventricular end-diastolic pressure of less than 15 mmHg at rest. Its fatal evolution brings patients to death, due to right ventricular failure in an average time of three years [1]. PAH incidence is estimated in 2.4-8 patients per million/year and its prevalence is 15-30 patients per million [2, 3].
The majority of PAH are idiopathic, while the rest may be: familial, associated with collagen diseases, congenital systemic-to-pulmonary shunts, portal hypertension, HIV infection, drugs and toxins [anorexigens, fenfluramine, rapeseed oil] and persistent pulmonary hypertension of the newborn [4].
PAH is a disease with a complex and not completely understood pathophysiology. Several pathophysiological pathways seem to be involved: prostaglandin pathway, acting on AMPc generation that can be fostered by the administration of prostacycline analogues [epoprostenol, treprostinil, iloprost, beraprost]; nitric oxide pathway, enhancing the production of GMPc, which is inhibited by phosphodiesterase-V that can be blocked by its inhibitors [sidenafil, tadalafil]; and the endothelin-1 pathway, acting as a potent endothelial vasoconstrictor that can be partially blocked by endothelin receptor antagonists [bosentan, ambrisentan, sitaxentan] [5]. New physiophatologic pathways including angiogenesis, TGF-β, serotonin, and cellular proliferation mechanisms are coming out and appear to be relevant in vascular structural changes related to PAH [6].
HISTOPATHOLOGY AND VASCULAR REMODELLING
Histopathology of PAH is founded on structural modifications on the vascular wall of small pulmonary arteries characterized by thickening of all its layers. These changes named as vascular remodelling include vascular proliferation, fibrosis, and vessel obstruction. Development of PAH involves the complex interaction of multiple vascular effectors at all anatomic levels of the arteriolar wall. Subsequent vasoconstriction, thrombosis, and inflammation ensue, leading to vessel wall remodelling and cellular hyperproliferation as hallmarks of disease severity. These processes are influenced by genetic predisposition as well as endogenous and exogenous stimuli. The final result of this process is that arterial vascular wall becomes thicker and more rigid modifying its behaviour in terms of elasticity, pulsatility and compliance.
Nowadays triggers of pulmonary vascular remodelling remain unknown. However it has been suggested that some mesenquimal undifferentiated cells placed on the vascular divisions where muscular smooth cell layer begins to disappear, may differentiate into smooth muscle cells due to exogenous stimuli, not recognized yet. A second hypothesis that would explain why myofibroblasts are the predominant cells in the hypertrophied muscular layer in PAH suggest that interstitial and adventitial fibroblasts, once activated, can differentiate into myofibroblasts that would migrate to the muscular layer. It has also been proposed that bone marrow stem cells could play a role in this process. When lung suffers an aggression or an inflammation mediated by cytokines, there is a release of bone marrow progenitor cells with the capability of differentiate in endothelial cells that would repair the damaged zone. There is some research in mice which demonstrates the migration of stem cells to the vessel wall if the animal is under hypoxia, showing that these cells could then proliferate and differentiate into smooth muscle cells [7-11]. Finally, there is the endothelial-to-mesenchymal transition theory, imported from the pathogeny of cancer and based on the endothelium ability of change into a mesenquimal tissue composed by smooth muscle cells after its stimulation by aggressions and growth and transforming factors [12]. A common phenomenon already demonstrated whatever the ethiology of the disease is, is that affected vessels show a lesser number of smooth muscle and intimal apoptotic cells, deed that favours indirectly the process of hyperplasia and hypertrophy of these cells [13].
Intimal Lesions
Intimal lesions largely influence the overall pulmonary vascular resistance. There are three kinds of lesions responsible of intimal remodelling, which appear sequentially along disease progression. The initial lesion consists on abnormal vasculogenesis leading to plexiform and angiomatoid lesions, typically located on vascular division points. This event shares characteristics and etiologic factors with some neoplasia, since absence of antiremodelating mediators and tumoral suppressor factors have been observed [13-15]. Plexiform lesions progress into “onion skin” lesions developed from intimal myofibroblasts and endothelial cells, both growing uncontrolled in concentric layers reducing significantly the luminal area [16]. The final stage of this process consists on accumulation of scarring extracellular matrix that contributes to occlusion phenomenon.
Medial Lesions
Medial thickening plays an important role in PAH pathogenesis, though a relationship between the degree of remodelling and the severity of the disease or the response to treatment has not been clearly demonstrated yet. Two factors have been suggested as triggers for medial remodelling: mutations in bone morphogenetic protein receptor 2 [BMPR2], implicated in the genesis of the disease [17] and vascular smooth muscle cells in PAH expressing higher levels of serotonin that promotes vasoconstriction, cell growth and enhances hypoxia induced remodelling [18]. Medial changes involve muscularized arteries and precapillary vessels where medial layer thickening is due to a combination of smooth muscle cells hypertrophy, and hyperplasia with a deposition of extracellular matrix and thickening of elastic laminae [19].
Adventitial Lesions
Adventitial thickening is mostly due to collagen deposition but fibroblasts also contribute with its sensitive response to stimulus like hypoxia or increased vascular pressure. This induces, due to their biological heterogeneity, differentiation into smooth muscle cells that migrate to medial layer, stimulation medial smooth muscle cells proliferation by secretion of growth factors, allowing the recruitment of inflammatory and bone marrow progenitors cells and creating a vasculogenic niche for the expansion of newly formed vessels [20]. All these changes promote development of PAH.
As already previously pointed in the introduction, death in PAH is usually due to terminal right cardiac failure.
Right ventricle is the main victim of the hemodynamic consequences of PAH. The adaptative fight against chronically elevated pulmonary vascular resistances resistances induces structural changes in the right ventricle that progressively leads to its functional impairment. Because of this, a renew interest in studying right ventricle in PAH has arisen nowadays. Doppler echocardiographic technology appears to be the most useful technique for the non invasive evaluation of right ventricular function in the clinical setting.
Hitherto, our knowledge about vascular remodelling mainly comes from autopsy studies as lung tissue is not usually available during the course of the disease. This fact has hindered all the investigations in this field but lastly, new vascular exploratory technologies like intravascular ultrasound (IVUS) or optical coherence tomography have arisen as potentially practical tools for the in vivo study of vascular changes in PAH.
Along this article we will go into exploratory techniques for evaluating vascular and cardiac remodelling in PAH focusing on new invasive techniques and advances in echocardiography.
INVASIVE AND NON INVASIVE EVALUATION OF PULMONARY ARTERIAL WALL AND RIGHT VENTRICLE
In clinical practice the diagnosis of Pulmonary Hypertension relies on measurements of pulmonary vascular pressure and cardiac output, and calculation of pulmonary vascular resistances [21]. These hemodynamic measurements are of prognostic value, with survival being related to cardiac output rather than to pulmonary pressure. Clinical signs of right ventricular failure often are not clearly related to progression of pulmonary hypertension as assessed by pulmonary pressure and resistance. This observation may be explained by the fact that heart failure in pulmonary hypertension is the consequence of both global changes of right ventricular overload, not only in pulmonary vascular resistances, and the consequence of uncoupling of right ventricle to the hypertensive pulmonary circulation, which is not actually measured by routine hemodynamic evaluation [22].
Pulmonary Arterial Wall
Direct evaluation of pulmonary vascular structure is not routinely performed in pulmonary hypertension since current imaging techniques are limited and since little is known about the relationship between structural changes and functional characteristics of the pulmonary vasculature. Intravascular ultrasound studies in patients with pulmonary hypertension have shown a thicker middle layer, increased wall-thickness ratio (Fig. 1) and diminished pulsatility than in control patients [23,24]. However vasoreactivity to nitric oxyde, oxygen or epoprostenol does not appear to correlate with structural assessment of pulmonary vascular wall by intravascular ultrasound (IVUS) [23, 24]. IVUS provides combined morphological and functional evaluation of the elastic pulmonary vessels of patients with pulmonary hypertension and allows direct assessment of the acute changes induced in the pulmonary vessel wall dynamics by epoprostenol. The exploration is safe, and can be undertaken at the same time of cardiac catheterization. In addition there is an association between impaired pulmonary artery functional state as determined by IVUS and mortality at follow-up. However larger series are needed to determine the exact prognostic value of this technique in the setting of pulmonary hypertension [23].
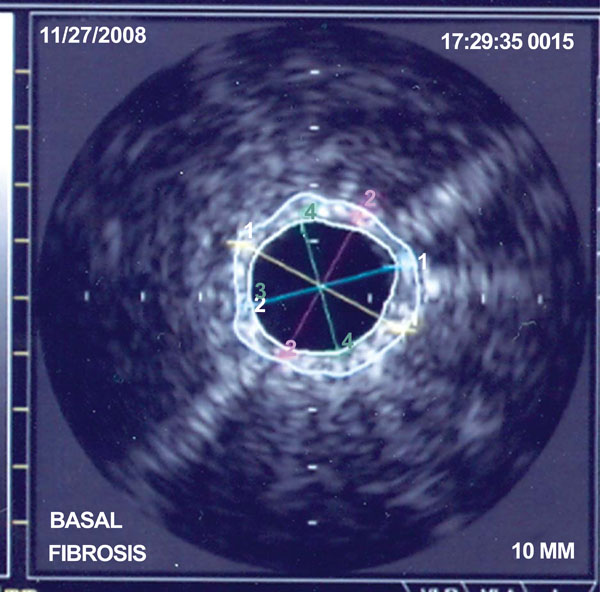
Image of IVUS of a pulmonary artery in a patient with Pulmonary Arterial Hypertension. Calculation of amount of fibrosis of the arterial wall.
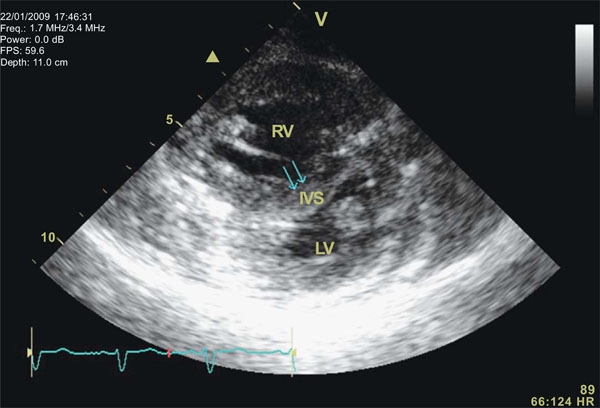
Short axis view of the right [upper] and left ventricle in a patient with PAH. It can be seen important septal shift [arrows] at the beginning of diastole that added to the right ventricle enlargement contributes to an important increase in the eccentricity index (quantifying the spherical shape of the right ventricle). RV: right ventricle. LV: left ventricle. IVS: interventricular septum.
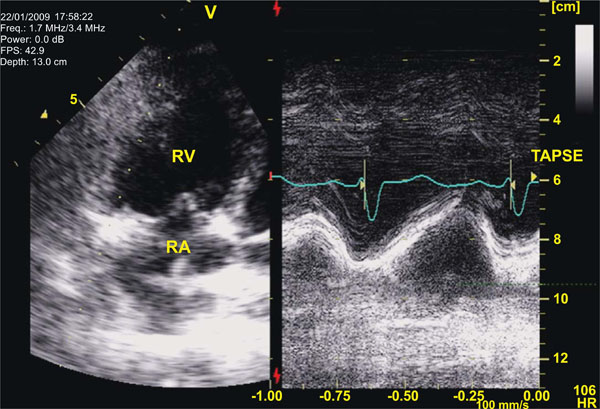
Tricuspid Annular Plane Systolic Excursion [TAPSE] in a patient with severe PAH during a vasodilatory test with epoprostenol: Left panel depicts the localization of the cursor line over the lateral portion of the tricuspid annulus on an apical view of the right heart chambers [RV: right ventricle; RA: right atrium]. On the right panel the M-mode represents displacement of the annular plane during the cardiac cycle. Main advantages of TAPSE, despite its dependency on volumetric assumptions, are that it is very easy to obtain and quite reproducible.
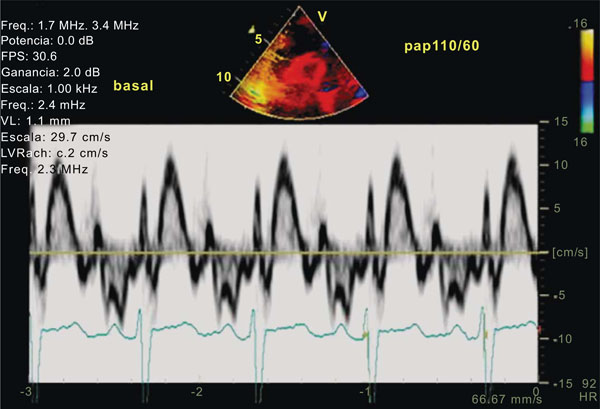
Tissue Doppler Imaging [TDI] of the Right Ventricle [RV]: Pulsed Wave Doppler as depicted in the image allows the analysis of regional velocities and measurement of both peak systolic and isovolumic acceleration [IVA] at different levels of the RV free wall. These parameters although not completely independent of loading conditions may offer information about RV systolic function that at least does not rely on geometric or volumetric assumptions.
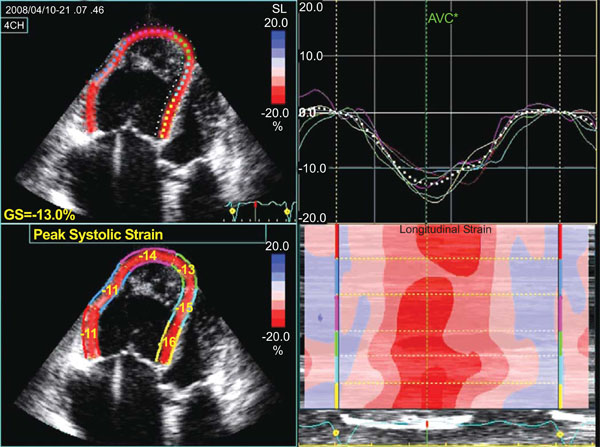
2D Strain analysis of the RV in a patient with severe PAH during a vasodilatory test. Measurement of deformation parameters of the RV, as in this case employing longitudinal strain, appears as a very attractive alternative for the evaluation of systolic function. However, validation of these parameters in large populations of patients with PAH is desirable before its use at the clinical routine. Left panel shows the measurements in the regions of interest (free wall and septum). Right upper panel shows the quantitative curves of longitudinal strain over time. Right lower panel shows the parametric quantitation in a colour scale of the different segments analysed.
It has to be taken into account that pulmonary arteries, in contrast to systemic arteries, have a much thinner smooth muscle layer under normal conditions, consistent with a low pressure system, and that, as an approximation, the pulmonary vascular resistance is divided equally between arteries, capillaries and veins [25]. In addition the pulmonary circulation is a low pressure, low resistance, highly distensible system. Recently it has been shown that total compliance of the pulmonary vascular bed, defined as the ratio of stroke volume to pulse pressure, predicts mortality in patients with pulmonary arterial hypertension [26]. A large part of total vascular bed compliance is located in the proximal arterial branches. Therefore the predictive value of pulmonary vascular compliance in chronic pulmonary arterial hypertension also pertains to increased stiffness of the proximal pulmonary arteries.
Pulmonary arterial hypertension is basically a disease of small distal pulmonary arteries, leading to an increase in pulmonary vascular resistances, and as a result, pulmonary arterial pressure. Increased pressure causes distension and stiffening of the proximal pulmonary artery, causing vessel wall remodelling, which in itself may influence stiffness.
As already known pulmonary arterial hypertension can have a variety of different causes, but all causes have an increased right ventricular afterload in common. Because this increased afterload may lead to ventricular hypertrophy, right ventricular failure and ultimately death, it is important to insure our understanding of the right ventricular afterload. Most often in the clinical setting this afterload is defined as pulmonary vascular resistance. Because pulmonary vascular resistance is the ratio of mean pressure and mean flow, it reflects the arterial load to a steady flow. However blood flow is pulsatile, and thus a complete description of ventricular afterload should also include the load to a pulsatile flow. A decreased arterial compliance, for example, may be of similar importance for ventricular afterload as an increased pulmonary vascular resistance [27].
Another determinant of right ventricular afterload that is neither reflected by resistance or compliance is the characteristic impedance of the proximal pulmonary arteries, which combines both blood inertia and vascular storage properties. A model that combines the 3 components of ventricular afterload is the 3 element windkessel model [28]. However there is not much information of the application of this model in the analysis of the pulmonary circulation.
These considerations on the different elements of the circulation contributing to right ventricular afterload may explain why pulmonary vascular resistance is a poor predictor of clinical prognosis in pulmonary arterial hypertension [22]. In addition in the pulmonary circulation the pulsatile components of pressure and flow contribute much as one third to one half of the total hydraulic power that is transferred from the right ventricle to the pulmonary vascular bed. As a consequence pulsatility plays a more important role in the pulmonary than in the systemic circulation. Pulsatility and mismatching of the right ventricle to its afterload may therefore better explain signs of clinical worsening than variables related to steady flow such as pressure and resistance [22, 23]. Therefore since pulmonary arterial compliance measures how much the aggregate pulmonary arterial tree will dilate with each contraction of the right ventricle, it should be inversely proportional to the workload on the right heart, and thus compliance will be more important than resistance in the long run right ventricular remodelling, and consequently a better predictor of prognosis and mortality in pulmonary arterial hypertension [23].
As already mentioned IVUS has been validated as a reliable method for describing pulmonary vessel wall morphology [23-30]. Recently Optical Coherence Tomography (OCT) a new high resolution imaging modality has proven its superiority over IVUS for the detection and characterization of coronary atherosclerotic plaque composition, specifically for the differentiation of non-calcified, lipid rich, or fibrous plaque [31]. However, at the moment no literature exists on the use of OCT in primary pulmonary hypertension. Optical Coherence Tomography is based on the measurement of the back-reflection of infra-red light. The principle of OCT has been described by other groups [32, 33].
The most important advantage of OCT over other intravascular imaging modalities is its superior resolution, that will probably allow a more precise quantification of the arterial wall fibrosis.
Right Ventricle
Clinical signs of right ventricular failure vary greatly in the presence of similar severity of pulmonary arterial pressure. Pulmonary arterial pressure and pulmonary vascular resistance are not sufficient to evaluate right ventricular afterload because this composite variable is flow dependent and because it does not integrate changes in arterial elastance and wave reflection. As the clinical consequences of pulmonary hypertension relate to the ability of right ventricular function to maintain adapted flow output, quantifying ventricular-arterial coupling appears to be a more logical hemodynamic approach [34]. Although this approach basically requires measurements of instantaneous right ventricular volumes, bedside application will obviously require a more simplified and non invasive approach. Current progress in Echo Doppler technology will probably make this possible allowing for point by point calculation of right ventricular pressure curves from the envelop of tricuspid regurgitant jets and synchronized measurement of instantaneous pulmonary arterial flow [35,36].
Methods for assessing right ventricular function include echocardiography, MRI, intermittent hemodynamic monitoring, implantable hemodynamic monitors, nuclear medicine, natriuretic peptide levels as well as indirect measures such as cardiopulmonary exercise testing. In the clinical setting echocardiography is clearly the most used and reliable technique.
The presence of pericardial effusion has long been recognized as an end-stage prognostic finding, occurring when the right heart fails. The degree of septal shift in diastole, and its correlate the eccentricity index, is also related to outcome and to improvement with effective therapy [37] (Fig. 2). However, these parameters depict ventricular failure, but do not directly assess right ventricular function.
The majority of the proposed methods of echocardiographic assessment of right ventricular function are based on volumetric approximations of the right ventricle. However such approaches have intrinsic limitations, first since volume related measures such as ejection fraction are load dependent, second because of the complex geometry of the right ventricle [38]. The issue of right ventricular geometry is usually overcome using the so called geometry independent parameters such as tricuspid annular velocity and tricuspid annular plane systolic excursion (TAPSE) [39] (Fig. 3) and three dimensional echocardiography [40]. Nevertheless assessments based on tricuspid annular excursion have important inherent limitations as independent measurements of right ventricular pumping function [41]. However, is it really necessary to analyse right ventricular volumes in order to calculate a variable so dependent on post load such as ejection fraction, particularly in a clinical setting where afterload is so important, such as pulmonary hypertension? It appears that the Echo Doppler technology will be more logically applied to the study of functional aspects unable to be quantified by other imaging techniques.
Another promising tool for right ventricular assessment is tissue doppler imaging techniques, but at present, data are relatively scarce and variability of results are still too high to rely only in this method for right ventricular function analysis in pulmonary hypertension (Fig. 4). However some studies have already reported normal range of mechanical variables in Doppler imaging studies [42], have shown evidence of interventricular dyssynchrony in pulmonary hypertension [43] and have related these variables to serum levels of brain natriuretic peptide [44]. In addition even some variables, such as apical strain rate (Fig. 5), have been correlated with pulmonary artery pressures [45].
CONCLUSIONS
We firmly believe that the remodelling process of the pulmonary vasculature and the right ventricle play an important role in the evolution of PAH and thus must be part of the evaluation of the disease progression and of the response to pharmacological treatments. Techniques such as IVUS, OCT and new Echo Doppler technology will probably allow to study in vivo this remodelling process.

