All published articles of this journal are available on ScienceDirect.

The Role of Sphingolipids and Ceramide in Pulmonary Inflammation in Cystic Fibrosis
Authors Info & Affiliations
Abstract
Sphingolipids and in particular ceramide have been shown to be critically involved in the response to many receptor-mediated, but also receptor-independent, mainly stress stimuli. Recent studies demonstrate that ceramide plays an important role in the pathogenesis of cystic fibrosis, a hereditary metabolic disorder caused by mutations of the Cystic Fibrosis Transmembrane Conductance Regulator. Patients with cystic fibrosis suffer from chronic pulmonary inflammation and microbial lung infections, in particular with Pseudomonas aeruginosa. Chronic pulmonary inflammation in these patients seems to be the initial pathophysiological event. Inflammation may finally result in the high infection susceptibility of these patients, fibrosis and loss of lung function. Recent studies demonstrated that ceramide accumulates in lungs of cystic fibrosis mice and causes age-dependent pulmonary inflammation as indicated by accumulation of neutrophils and macrophages in the lung and increased pulmonary concentrations of Interleukins 1 and 8, death of bronchial epithelial cells, deposition of DNA in bronchi and high susceptibility to Pseudomonas aeruginosa infections. Genetic or pharmacological inhibition of the acid sphingomyelinase blocks excessive ceramide production in lungs of cystic fibrosis mice and corrects pathological lung findings. First clinical studies confirm that inhibition of the acid sphingomyelinase with small molecules might be a novel strategy to treat patients with cystic fibrosis.
CYSTIC FIBROSIS
Cystic fibrosis (CF) is caused by mutations of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) [1-3] and constitutes with 1 case per 2500 births the most common autosomal recessive disorder in the EU and the USA. Approximately 40 000 children and young adults are affected in the EU. The genetic defect of the CFTR-molecule results in several clinical symptoms, in particular pulmonary and gastrointestinal problems. Gastrointestinal symptoms include a defective secretion of pancreatic enzymes and alterations of the liver, pulmonary problems are in particular recurrent and chronic infections with Pseudomonas aeruginosa (P. aeruginosa) and Burkholderia cepacia, but also Staphylococcus aureus and Haemophilus influenzae. The cause for the high sensitivity of cystic fibrosis patients to develop these pulmonary infections is unknown.
P. aeruginosa infections are most important in patients with cystic fibrosis. Once past early childhood a very high percentage of patients with cystic fibrosis suffers from recurrent and finally chronic P. aeruginosa pneumonia [4]. More than 80% of adult patients with cystic fibrosis are infected with the pathogen and chronic P. aeruginosa lung infections are the leading cause of morbidity and mortality in cystic fibrosis [4].
INFLAMMATION IN CYSTIC FIBROSIS
Since CFTR exhibits a chloride channel activity, it was speculated that water absorption in the mucus present on epithelial cells of the respiratory tract is altered, which may result in a reduced mucociliary clearance and a reduced ability to eliminate P. aeruginosa [5, 6]. The increased viscosity of the mucus may also affect the ability of neutrophils to migrate to and kill bacteria in the respiratory tract. However, although in vitro experiments suggested the concept of reduced mucociliary clearance in cystic fibrosis [5], in vivo studies on cystic fibrosis patients failed to demonstrate a significant and uniform reduction of the mucociliary clearance in these individuals [6].
A defect in the chloride channel function of CFTR may also result in changes of the ion concentration in the mucus. The altered concentration of ions may then impair the function of defensins and other anti-microbial peptides and, thus, reduce the elimination of P. aeruginosa in the lung [7]. However, at present it is unknown whether the defect of CFTR really results in hypertonic salt concentrations on the respiratory epithelium in vivo and, thus, dysfunction of antimicrobial peptides.
Several studies suggest a defect of vesicular pH-regulation in cystic fibrosis. Barasch et al. were first to show that CFTR participates in pH control of trans-Golgi vesicles [8]. A defect of CFTR resulted in alkalinization of these vesicles. More recently, it was shown that Cftr contributes to acidification of phagolysosomes in macrophages [9]. Cftr seems to control the influx of chloride ions into lysosomes, which serve as counterions for protons pumped into phagolysosomes by the lysosomal V-type H+-ATPase. Deficiency of Cftr limits accumulation of protons in phagolysosomes and, thus, acidification of these vesicles to pH values less than 5-6. The defective acidification of phagolysosomes, in particular in Cftr-deficient alveolar macrophages, blocks the bactericidal activities of lysosomal enzymes and, thus, the ability of macrophages to kill internalized bacteria [9]. Recent studies from our laboratory on freshly isolated epithelial cells from Cftr-deficient mice confirmed previous studies by Di et al. that Cftr controls the pH of at least some vesicle populations in these cells [10]. Further unpublished studies from our group using freshly isolated macrophages from Cftr-deficient mice demonstrate that Cftr controls the pH in a specific vesicle population, most likely secretory lysosomes, already in non-infected cells, while the pH in endocytotic vesicles is independent of Cftr. The specific role of Cftr in the control of the pH of a distinct subset of vesicles also explains findings of other groups [11, 12] that reported that Cftr-deficiency does not control lysosomal pH. These studies investigated the pH in vesicles employing zymosan-conjugates containing fluorescein and tetramethylrhodamine-dextran and, thus, these studies report the pH in vesicles that were generated upon endocytosis of these dyes [11, 12]. This suggests that Cftr only determines the pH in distinct vesicle populations, most likely pre-lysosomes, secretory lysosomes and lysosomes that are not part of the endocytosis machinery.
Pier et al. demonstrated a defect of the innate immune function of epithelial cells in the respiratory tract of cystic fibrosis mice. They showed that the lipopolysaccharide (LPS)-molecule of P. aeruginosa binds to a short amino acid sequence in CFTR (AA 103-117) [13]. This interaction mediates internalisation of P. aeruginosa into lung epithelial cells. Deficiency of Cftr or blockade of the binding sequence prevents P. aeruginosa internalisation [13]. Bacterial internalisation correlated with the defense against P. aeruginosa in lungs of infected mice, although the causative role of the internalisation defect for the high sensitivity of these mice to develop P. aeruginosa infections remains to be demonstrated. Further studies suggested that internalisation of P. aeruginosa is also mediated by other mechanisms. The uptake mechanisms also seem to depend on the bacterial strain, since rough, mucoid strains of P. aeruginosa are able to enter epithelial cells independently of CFTR expression [14].
Studies by several groups demonstrate that P. aeruginosa triggers cell death of Cftr-positive epithelial cells in vitro as well as in a focal pattern in bronchial epithelial cells in the lung. Apoptosis is mediated by an activation of the endogenous CD95 receptor/CD95 ligand-system within distinct membrane domains in epithelial cells upon infection with P. aeruginosa [15-18]. In vivo studies indicate that stimulation of CD95 and, thus, presumably induction of apoptosis in epithelial cells is central for a coordinated defense against pulmonary P. aeruginosa infections. It might be possible that the induction of apoptosis in epithelial cells modulates the local immune response to the pathogen and prevents an over-exaggerated release of cytokines [19, 20]. Studies from Kumar et al. on two independent CF patient cohorts demonstrate that allelic variants within intron 2 of the CD95 gene modulate the manifestation of CF disease further suggesting a role of CD95-mediated signalling/ apoptosis in cystic fibrosis [21].
Several studies in the recent years suggest an imbalance between pro-inflammatory and anti-inflammatory cytokines in the airways of cystic fibrosis patients as one of the leading causes to result in destruction of the lung and the high susceptibility of these patients to infections with P. aeruginosa. It was demonstrated that cells lacking functional CFTR secrete increased amounts of Interleukin (IL)-1, IL-8/KC, TNF-alpha and Mip-2 upon infection with P. aeruginosa compared to control cells [22-25]. IL-8 is already increased in the trachea of non-infected Cftr-deficient mice [23], while the synthesis of anti-inflammatory cytokines, in particular IL-10 [25], has been shown to be reduced in Cftr-deficient mice under basal conditions as well as after infection with P. aeruginosa. The increased release of proinflammatory cytokines from CFTR-deficient epithelial cells correlates with a constitutive activation of NFκB in these cells [26, 27]. Several studies indicated that at least some mutations of Cftr result in accumulation of the mutated protein in CF cells, which triggers endoplasmic reticulum stress and finally increased NFκB-activity [28]. In addition, a recent study indicated that Cftr must be present in membrane rafts to suppress activation of NFκB in CF cells [29] suggesting that Cftr controls several checkpoints of NFκB-activity.
CERAMIDE
The cell membrane contains sphingolipids, cholesterol and (glycero)phospholipids among other less abundant lipids. Sphingolipids consist of a hydrophobic ceramide moiety and a hydrophilic headgroup. Ceramide is composed of D-erythro-sphingosine and a fatty acid containing 2-28 carbon atoms in the acyl chain that is connected via an amide ester bond [30]. Based on biophysical experiments determining the melting temperatures of lipids, Singer and Nicolson introduced the classical fluid mosaic model in 1972 predicting free movement of proteins in the lipid bilayer [31]. However, this concept was revised in the last 10-15 years and present concepts indicate the existence of distinct membrane domains [32, 33]. Sphingolipids interact with each other and with cholesterol molecules via hydrophilic interactions between the sphingolipid headgroups and the hydroxy group in the cholesterol molecule, respectively, and via hydrophobic van der Waal interactions between ceramide moieties and the sterol ring system [32-35]. The tight interactions of sphingolipids and cholesterol were suggested to promote the transition of membrane lipids into a liquid ordered status and the formation of very small distinct domains in the cell membrane, named rafts [32]. Rafts are proposed to be very small membrane domains enriched in sphingolipids and cholesterol that spontaneously separate from other phospholipids in the cell membrane [32-35]. Recent microscopy studies demonstrated rafts in cells and indicated a size of approximately 20 nm [35]. These experiments also suggest that rafts serve to sort proteins in the cell membrane and, thus, to contribute to a lateral organization of the cell membrane.
The generation of ceramide within rafts, but also independent of rafts, dramatically alters membrane properties. Ceramide molecules self-associate to small ceramide-enriched membrane microdomains, which have the tendency to spontaneously fuse to large ceramide-enriched membrane domains [36-38]. The formation of ceramide-enriched membrane domains was shown to occur in cells after stimulation via a variety of stimuli including CD95 [39-41], CD40 [42], DR5 [43], FcγRII [44], the PAF-receptor [45], but also after infection with P. aeruginosa [19], Neisseriae gonorrhoeae [46], Rhinovirus [47], application of stress stimuli such as UV-light [48], cisplatin [49] or Cu2+-treatment [50]. The great variety of stimuli that trigger the formation of ceramide-enriched membrane platforms suggests that these membrane domains facilitate signal transduction, but are very likely not part of the specific signal transduction pathway elicited by the these stimuli.
We suggested that ceramide-enriched membrane domains serve to re-organize receptor molecules and intracellular signaling molecules in the cell membane [51], which permits the specific receptor to transmit its signal into the cell. Ceramide-enriched membrane domains exhibit very different biophysical properties than other parts of the plasma membrane resulting in preferential trapping of activated receptor molecules and/or signaling molecules in these domains. The trapping of proteins within ceramide-enriched membrane domains might be enhanced by a direct interaction of ceramide with proteins. However, at present the molecular details of receptor clustering and preferential sorting of molecules to ceramide-enriched membrane domains are unknown. It was shown that the transmembrane domain of CD40 is critically involved in trapping of the receptor to ceramide-enriched domains [52]. Thus, an alteration of the transmembrane domain upon binding of the ligand to its receptor may provide specificity for protein-sorting into ceramide-enriched membrane domains. Clustering of receptors and associated signaling molecules may result in a density of receptors and signaling molecules, the spatial association of activated receptors with intracellular molecules, the exclusion of inhibitory enzymes and/or transactivation of intracellular enzymes. Thus, clustering might coordinate the spatial and temporal organization of the signalosome generated by a specific receptor.
In addition, ceramide has been shown to directly regulate several molecules including cathepsin D [53], phospholipase A2 [54], kinase suppressor of Ras [55], ceramide-activated protein serine-threonine phosphatases (CAPP) [56] and protein kinase C isoforms [57, 58]. Further, ceramide has been shown to regulate the activity of the potassium channel Kv1.3 and calcium release-activated calcium (CRAC) channels [59-60]. Ceramide inhibits the activity of these ion channels in the cell membrane [59-62]. At present it is unknown how ceramide affects the activity of ion channels, but it might be possible that ceramide alters ion channels by a direct interference or by the change of the lipid composition in the environment of the channel [62]. In addition, studies by Siskind and Columbini demonstrated that ceramide molecules form channels in the outer mitochondrial membrane, which might contribute to the induction of apoptosis [63].
Many of these signalling molecules directly affected by ceramide are also good candidates to be involved in the regulation of inflammation by ceramide, for instance cathepsin D, phospholipase A2, kinase suppressor of Ras, Kv1.3 and calcium release-activated calcium channels. The role of ceramide in cystic fibrosis is described below.
SPHINGOMYELINASES
Ceramide is generated by (i) sphingomyelinases [30, 64, 65], (ii) by a de novo synthase pathway [30, 65], (iii) by a retrograde activity of the enzyme ceramidase that mediates the synthesis of ceramide from sphingosine [66], (iv) by the hydrolysis of complex glycosylated lipids [67] and (v) by hydrolysis of ceramide 1-phosphate [68]. Sphingomyelinases hydrolyse sphingomyelin to release ceramide. Sphingomyelinases display peak activities depending on the pH and, thus, were named acid, neutral and alkaline sphingomyelinase. Mammalian cells express one gene for the acid sphingomyelinase, which can be targeted to lysosomes or for secretion into secretory lysosomes depending on the glycosylation pattern of the protein [69, 70]. Mammalian cells express at least two different neutral sphingomyelinases and one alkaline sphingomyelinase [65].
The molecular mechanisms of the regulation of sphingomyelinases still require definition. The activity of neutral sphingomyelinases seems to be regulated by the redox balance in the cell [71]. However, further details of the stimulation of neutral sphingomyelinase are presently not known. The acid sphingomyelinase is activated by oxidation [43, 50, 72]. It is unknown whether the protein is directly oxidized in vivo or regulated via pathways that are redox-sensitive. Scavengers of reactive oxygen intermediates prevent activation of the acid sphingomyelinase by stimuli such as DR5 or Cu2+ [43, 50, 72]. In vitro studies by Qui et al. [72] on purified acid sphingomyelinase suggest a direct oxidation of the acid sphingomyelinase at cysteine residue 629 with a subsequent copper-promoted dimerisation of the acid sphingomyelinase [72]. The oxidation of cysteine 629 correlated with an activation of the enzyme in vitro. In addition, phosphorylation of the protein at serine 508 has been shown to activate the enzyme [73]. The enzyme is inhibited by inositol-phosphates [74, 75], although the physiological significance of these findings remains to be determined.
The acid sphingomyelinase has been shown to be activated within lysosomes or multilamellar bodies, respectively, for instance after cellular stimulation via TNF receptors [53]. Other receptors, for instance CD95 or DR5, or infections with P. aeruginosa trigger a translocation of the acid sphingomyelinase onto the extracellular leaflet [39, 40, 43]. Translocation of the enzyme onto the outer leaflet of the cell membrane is very likely mediated by the fusion of vesicles, in particular secretory lysosomes that contain the enzyme [76], with the plasma membrane. The mobilisation of these vesicles and the fusion with the cell membrane occurs within seconds after cellular stimulation or infection, respectively.
Several studies [77-80] identified pharmacological inhibitors of the acid sphingomyelinase; the acid sphingomyelinase seems to interact with intra-lysosomal membranes, thereby being protected against proteolytic inactivation [77]. Weak bases such as amitriptyline diffuse into cells, are protonated in lysosomes and concentrated in these organelles. High pKa- and high logP-values of the drugs are necessary but not sufficient to functionally inhibit the acid sphingomyelinase [80]. These drugs interfere with binding of the acid sphingomyelinase to the membrane, which results in a detachment of the acid sphingomyelinase and subsequent proteolytic degradation [77-80]. The lipophilic ring structures that are present in all acid sphingomyelinase inhibitors are embedded in the lysosomal membrane, while the cationic amino group linked through an aliphatic chain to the ring system of all acid sphingomyelinase inhibitors interferes with binding of the enzyme to the membrane finally resulting in detachment and subsequent proteolytic degradation in the lysosome [80]. Thus, weak bases are not attacking the active center of the acid sphingomyelinase, but functionally inhibit the acid sphingomyelinase. It should be noted that acid sphingomyelinase inhibitors usually act fast and for a prolonged time.
CERAMIDE IN CYSTIC FIBROSIS
We have recently demonstrated that ceramide plays a critical role in the development of cystic fibrosis and the high sensitivity of Cftr-deficient mice to P. aeruginosa infections [11, 20]. Employing biochemical, fluorescence microscopy and mass spectrometry techniques, we revealed an accumulation of ceramide in lungs, in particular in epithelial cells of large and small bronchi, in different Cftr-deficient mouse strains (Fig. 1). Of note, ceramide accumulates in these cells prior to any infection. Further, ceramide accumulates in the lungs of Cftr-deficient mice in an age-dependent manner, which seems to mimic the delayed onset of symptoms in humans. We suggested that an alteration of the pH particular in secretory lysosomes, prelysosomes and/or lysosomes causes the accumulation of ceramide in Cftr-deficient cells (Fig. 2). Since the signalling pool of the acid sphingomyelinase seems to be predominantly localized to secretory lysosomes, we favour a primary accumulation of ceramide within these vesicles, although this hypothesis has yet to be proved. As described above Cftr-deficiency results in alkalinization of some, but not all, intracellular vesicles, in respiratory epithelial cells and macrophages. It should be noted that other vesicles, in particular endosomes, with a different pattern of ion channel expression than secretory lysosomes may not be alkalinized in Cftr-deficient cells. Since the enzymes that control ceramide metabolism in acidic vesicles are pH-dependent, an alkalinization of these vesicles has a dramatic effect on ceramide concentrations; sphingomyelin is constitutively metabolized to ceramide by the acid sphingomyelinase and consumed to sphingosine by the activity of the acid ceramidase [30]. An increase of the pH from values around 4.5 to pH 6.0 reduces the activity of the acid sphingomyelinase by only ~30%, while the acid ceramidase is almost inactived at pH 6.0 [11]. The imbalance of the two enzymes activities with a relative over-activity of the acid sphingomyelinase may then result in an accumulation of ceramide.
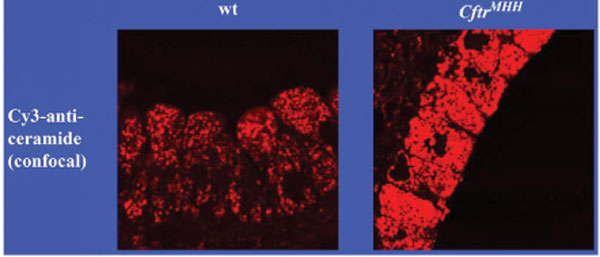
Accumulation of ceramide in bronchial epithelial cells of Cftr-deficient mice. The figure demonstrates the accumulation of ceramide in bronchial epithelial cells of Cftr-deficient mice. Lungs from wildtype (wt) or Cftr-deficient mice (CftrMHH) were removed, fixed in 4% paraformaldehyde and stained with Cy3-coupled anti-ceramide antibodies. The sections were analysed by fluorescence microscopy. The fluorescence intensity represents the concentration of ceramide in epithelial cells. Printed with permission of Nature Medicine [11].
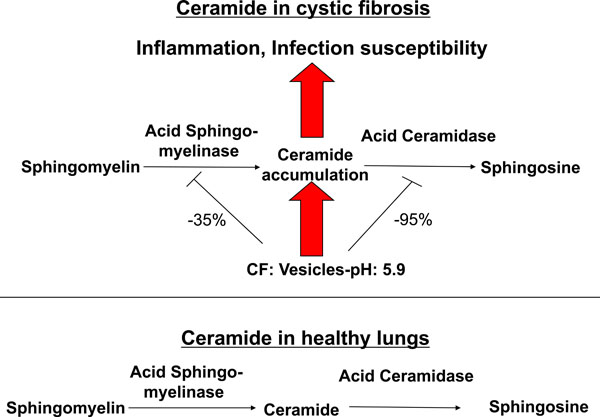
Effects of ceramide in cystic fibrosis. Alkalinization of intracellular vesicles, most likely secretory lysosomes, in CF-cells results in an imbalance of the activities of the acid sphingomyelinase and the acid ceramidase. The increase of the intravesicular pH to approximately pH 6 reduces the activity of the acid sphingomyelinase, which releases ceramide from sphingomyelin, by only 30-40%, while the activity of the acid ceramidase, which consumes ceramide, is reduced by more than 90%. Ceramide may then accumulate in CF cells as the net effect of the pH-mediated imbalance between the acid sphingomyelinase and the acid ceramidase. Ceramide induces cell death with subsequent deposition of DNA in the airways, chronic inflammation as indicated by accumulation of neutrophils, macrophages and proinflammatory cytokines such as Interleukin 1 and 8 in lung tissues and high susceptibility to P. aeruginosa infections. Genetic or pharmacological inhibition of the acid sphingomyelinase normalizes pulmonary ceramide, cell death, inflammation and infection susceptibility.
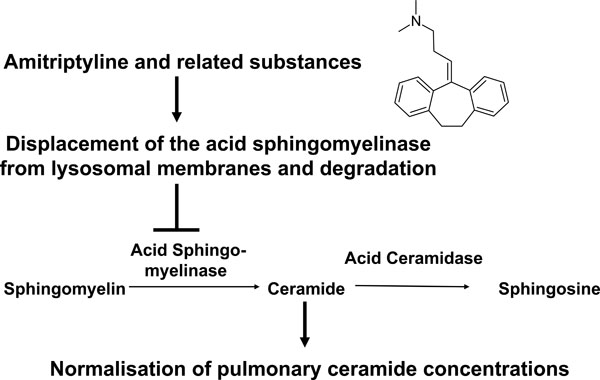
Model for the function of small molecule inhibitors of the acid sphingomyelinase in cystic fibrosis. Small molecule inhibitors of the acid sphingomyelinase as amitriptyline are weak bases that diffuse into acidic vesicles, are protonated and, thus, accumulate in these vesicles. The lipophilic ring structures of these molecules interact with the lysosomal membrane, while the cationic amino group on the aliphatic chain interferes with the binding of acid sphingomyelinase molecules to the membrane. This interference results in detachment of the acid sphingomyelinase from the lysosomal membrane, which leads to subsequent proteolytic degradation of the enzyme in the lysosomal lumen. The described inhibitors of the acid sphingomyelinase are, therefore, functional inhibitors of the enzyme. Application of functional inhibitors of the acid sphingoyelinase to Cftr-deficient mice results in reduction of ceramide release from sphingomyelin and, thus, normalization of pulmonary ceramide concentrations.
Pulmonary ceramide accumulation was observed in two strains of Cftr-deficient mice [11]. Ceramide accumulation was also observed in ciliated nasal epithelial cells and lung specimens from transplant material from patients with cystic fibrosis [11]. This suggests that the mouse data also apply to individuals with cystic fibrosis. Partial genetic or pharmacological inhibition of the acid sphingomyelinase almost normalizes ceramide concentrations in lungs of Cftr-deficient mice. Genetic inhibition of the acid sphingomyelinase was achieved by crossing Cftr-deficient mice with acid sphingomyelinase deficient mice (Smpd1-/- mice, Smpd1 is the gene symbol for sphingomyelinase phosphodiesterase 1, the systematic name for the acid sphingomyelinase) [11]. This resulted in mice deficient for Cftr and heterozygote for the acid sphingomyelinase in (Cftr-/-/Smpd1+/- mice) [11]. The activity of the acid sphingomyelinase in the lung of those mice was approximately 50% lower than in wild type mice [11]. Pharmacological inhibition of the acid sphingomyelinase was achieved by treatment of Cftr-deficient mice with the functional acid sphingomyelinase inhibitor amitriptyline [11].
Increased ceramide concentrations in bronchial epithelial cells and lung macrophages of Cftr-deficient mice trigger several hallmarks of cystic fibrosis, i.e. death of respiratory epithelial cells and deposition of DNA in bronchi, chronic pulmonary inflammation and high susceptibility of Cftr-deficient mice to develop pulmonary P. aeruginosa infections (Fig. 2).
The causative role of ceramide for increased cell death in the respiratory tract of Cftr-deficient mice is established by pharmacological and genetic inhibition of the acid sphingomyelinase in Cftr-deficient mice. Inhibition of ceramide accumulation in Cftr-deficient mice normalizes the increased death rates of bronchial epithelial cells in these mice. Ceramide has been demonstrated to mediate apoptosis by CD95 and DR5. Ceramide also activates cathepsin D in lysosomes to initiate the mitochondrial death pathway upon TNF-receptor activation [39, 43, 53]. However, at present it is unknown whether any death receptors or cathepsin D are involved in cell death observed in the airways of Cftr-deficient mice. Many studies have implicated short chain ceramides as triggers of apoptosis, however, they do not reflect the functions of physiological, long chain ceramides. Molecular mechanisms that mediate cell death by increased concentrations of cellular, long chain ceramides are largely unknown. Studies by R. Kolesnick et al. demonstrated the generation of mitochondrial ceramides upon irradiation, which triggers mitochondrial changes resulting in apoptosis [81]. However, it has to be determined whether a similar mechanism applies to effects of chronically accumulated ceramide in Cftr-deficient cells. Further, it remains to be defined which type of cell death, i.e. apoptosis, necrosis or any atypical form of cell death, is induced by ceramide in airway epithelial cells of Cftr-deficient mice. Finally, the signalling pathways that mediate ceramide-induced cell death in the respiratory tract of cystic fibrosis mice remain to be determined.
In any case, increased cell death in the respiratory tract of Cftr-deficient mice results in deposition of extracellular DNA on epithelial cells [11]. These DNA deposits may facilitate bacterial adherence in the respiratory tract and, thus, are critical for the development of the high infection susceptibility of cystic fibrosis mice to P. aeruginosa infections. This is supported by the findings that normalization of pulmonary ceramide concentrations upon treatment of cystic fibrosis mice with amitripytline or induced by acid sphingomyelinase heterozygosis in Cftr-/-/Smpd1+/- mice does not only prevent increased cell death and DNA deposition, but also normalizes the increased susceptibility of these mice to P. aeruginosa infections. Likewise, inhalation of DNase or inhibitors of the acid sphingomyelinase, respectively, reduces DNA-deposits in lungs of cystic fibrosis mice and prevents infection with P. aeruginosa [11, 82]. This suggests an important role of DNA-deposits in the airways of cystic fibrosis mice for the development of the high infection susceptibility of these mice to P. aeruginosa.
It is important to note that pulmonary inflammation elicited by ceramide in the respiratory tissue of Cftr-deficient mice preceed any bacterial infection. Again, partial inhibition of the acid sphingomyelinase by application of amitriptyline or in Cftr-/-/Smpd1+/- mice reduces the concentration of inflammatory cytokines in the lung and abrogates the accumulation of neutrophils and macrophages indicating the critical role of ceramide for the development of chronic inflammation in cystic fibrosis. Many lung functions are negatively affected by chronic inflammation, which may lead to a reduction of mucociliary clearance, local immune response and alterations of the airway architecture possibly resulting in pulmonary fibrosis.
At present it is unknown how increased ceramide concentrations mediate the upregulation of pro-inflammatory mediators and the accumulation of neutrophils and macrophages in the lung. It might be possible that enhanced cell turn over and induction of necrotic cell death trigger inflammation. However, it is also possible that other mechanisms such as increased expression of cytokines, alterations of cell migration, induced expression of adhesion molecules on endothelial cells to attract neutrophils and macrophages, etc. are important for these effects of ceramide.
Sphingolipids, in particular sphingomyelin, were recently shown to be a component of pulmonary surfactant [83]. Whether the alterations of pulmonary ceramide also affect surfactant in cystic fibrosis patients is presently unknown. A first hint pointing in this direction is the observation that local instillation of TNFα impairs surfactant functions and production that correlates with increased alveolar sphingomyelin levels, sphingomyelinase activity and possibly also ceramide concentrations [84].
In addition, the role of ceramide in the development of fibrosis, which is one of the major problems of children with cystic fibrosis, remains to be determined. Fibrosis does not seem to be a simple consequence of chronic infection, since fibrosis also occurs in Cftr-deficient mice without infection. It will be of great interest to investigate whether recent data that show a central role of ceramide for the development of fibrosis in chronic obstructive pulmonary disease also apply for cystic fibrosis.
P. AERUGINOSA INFECTIONS AND CERAMIDE
Previous studies published by our group indicate that P. aeruginosa infection of epithelial cells and macrophages activates the acid sphingomyelinase and triggers a translocation of the enzyme onto the extracellular leaflet of the cell membrane [19, 20]. The activity of the acid sphingomyelinase triggers the formation of ceramide-enriched membrane platforms in the outer leaflet. Activation of the acid sphingomyelinase is mediated by reactive oxygen species, at least in macrophages [20]. Recent studies demonstrate that activation of the acid sphingomyelinase is defective in cells lacking Cftr [85]. This defect might be caused by the altered pH in intracellular vesicles in Cftr-deficient cells, although the cause for the failure of P. aeruginosa to trigger acid sphingomyelinase and ceramide in Cftr-deficient cells remains to be determined. Ceramide-enriched membrane domains might serve the clustering of receptors, for instance CD95 and Cftr [19]. Genetic deficiency of the acid sphingomyelinase or disruption of rafts by interference with cellular cholesterol prevents formation of ceramide-enriched membrane platforms, internalisation of the bacteria and induction of apoptosis in epithelial cells upon infection with P. aeruginosa, but results in an uncontrolled cytokine release [19]. The mechanisms that mediate an uncontrolled release of cytokines in the lung of Smpd1-deficient mice infected with P. aeruginosa are presently not known.
Acute pulmonary infection of Smpd1-deficient mice with P. aeruginosa reveal that these mice are unable to remove the bacteria upon primary lung infection and a high percentage of the mice succumbed by a sepsis within a few days, while wild type mice are resistant [19]. These findings indicate that ceramide is required for the removal of the bacteria in the lung, at least during an acute infection. These data seem to be contradictory to the results discussed above in cystic fibrosis mice, since both, a defect to respond to the infection with an activation of the acid sphingomyelinase and a release of ceramide as well as a chronic over-production of ceramide negatively affect the capability of the lung to deal with P. aeruginosa. However, this situation seems to be typical for many important biological processes. For instance, an increased coagulation might result in infarcts, while a defect of blood coagulation results in spontaneous bleedings. Thus, only a moderate, therapeutic inhibition of blood coagulation suppresses infarcts and at the same time prevents bleedings. Further, the two situations are very different, since Smpd1-/- mice are unable to remove the bacteria from the lung once the infection has been established, while the initial number of bacteria in the lung of these mice does not differ from that in wildtype mice immediately after infection. In contrast, Cftr-deficient mice suffer from a defect in the early phase of the infection period, i.e. high numbers of bacteria reach the lung or are able to remain in the lung immediately after infection. The increased susceptibility to develop P. aeruginosa infections might be caused by induction of cell death and DNA depositions in bronchi that serve the adhesion of the bacteria [11]. The therapeutic implications of these considerations are discussed below.
INHIBITION OF THE ACID SPHINGOMYELINASE BY SMALL MOLECULE DRUGS TO TREAT CYSTIC FIBROSIS
The studies in mice indicate that increased ceramide concentrations are deleterious for cystic fibrosis patients and reduction of ceramide levels by targeting the acid sphingomyelinase might be beneficial. However, ceramide concentrations should not be reduced under a critical cellular level by any drug to preserve the biological functions of ceramide. Unpublished studies from our group demonstrate that already 5-15% residual activity of the acid sphingomyelinase permitted a normal response of epithelial cells to P. aeruginosa infection. Therefore, treatment with amitriptyline in doses that mediate an inhibition of 50-70% of the acid sphingomyelinase activity seems to be very safe and, at the same time, sufficient to normalize ceramide levels (~5 pmol/μg lung protein) in cystic fibrosis (Fig. 3).
Recently, results from a first randomised, double-blinded, placebo-controlled, cross-over pilot study on 4 adult CF patients and a phase IIa study on 19 adult CF patients were published [86]. These studies revealed that amitripytline is safe in patients with cystic fibrosis. They further demonstrate that even low doses of amitriptyline significantly increased lung functions in cystic fibrosis patients, which was measured as forced expiratory volume in the first second (FEV1). Finally, ceramide levels determined in respiratory epithelial cells from cystic fibrosis patients decreased significantly under amitriptyline therapy. Although the number of patients tested in these studies is still low, these data suggest amitriptyline and related substances as a novel therapeutic option in patients with cystic fibrosis.
However, these studies were performed with systemic application of amitriptyline, which results in some side effects, for instance dry mouth or tiredness [86]. Thus, an inhalation of the drugs will be definitely preferred over a systemic application to achieve higher local doses and, at the same time, to avoid any systemic effect. The latter applies in particular for treatment of children and for any long-term use. The inhalative application of drugs that inhibit the acid sphingomyelinase and locally reduce ceramide in lungs of cystic fibrosis patients must be developed and tested. However, the above-described studies in mice and first clinical data already identified this group of small molecules as potential drugs to treat cystic fibrosis and justify further development of these small molecule drugs.
ACKNOWLEDGEMENTS
Studies described in the present manuscript were supported by the AKF-program of the University of Tuebingen to J.R. and DFG grants Gu 335-16/1 and Gu 335-20/1 to E.G.

