All published articles of this journal are available on ScienceDirect.

Safety and Efficacy of Fluticasone Propionate/Salmeterol Hydrofluoroalkane 134a Metered-Dose-Inhaler Compared with Fluticasone Propionate/Salmeterol Diskus in Patients with Chronic Obstructive Pulmonary Disease
Authors Info & Affiliations
Abstract
Purpose: To provide information on the efficacy and safety of Fluticasone Propionate/Salmeterol Hydrofluoroalkane 134a Metered-Dose-Inhaler 230/42mcg (FSC MDI) and its comparable dose of Fluticasone Propionate/Salmeterol DISKUS 250/50mcg (FSC DISKUS) in patients with COPD.
Methods: This multicenter, randomized, double-blind, 12 week study was designed to evaluate FSC MDI treatment responses as compared with FSC DISKUS. The primary comparison of interest was non-inferiority between the FSC MDI treatment group and the FSC DISKUS treatment group assessed in terms of 2-hour post-dose FEV1 change from baseline at endpoint. The non-inferiority criterion bound was 75mL (lower confidence limit of -75mL). Inclusion criteria: Male or female aged ≥ 40, post-bronchodilator FEV1 ≤ 70% predicted normal, FEV1/FVC ≤ 70% and ≥ 10 pack years smoking history. Adverse events were recorded by patients throughout the study on daily diary cards. Adverse events were collected in eCRFs at all clinic visits and during a final follow-up phone call.
Results: Patients (N=247) were randomized to FSC MDI (FEV1% 49.3 + 12.3, FEV1/FVC 50.5 + 10.0) and FSC DISKUS (FEV1% 48.4 + 11.0, FEV1/FVC 50.3 + 10.3). From an ANCOVA model the least squares (LS) mean difference (FSC MDI– FSC DISKUS) for the 2-hour post dose FEV1 at endpoint was -2.0mL (95% CI -64mL, 59mL). Pre-dose FEV1, FVC, PEF, and albuterol use were also similar between the two formulations. The most common adverse events (AE) during treatment were headache (8% and 6% of patients), nasopharyngitis (4% and 6%), cough (3% and 4%), and sinusitis (2% and 5%) for FSC MDI and FSC DISKUS, respectively. Pneumonia was recorded as an AE for 2 (2%) patients in the FSC DISKUS arm.
Conclusion: This is the first study to demonstrate that FSC MDI has a similar efficacy and safety profile to FSC DISKUS in COPD patients.
INTRODUCTION
Current evidence-based COPD guidelines recommend the use of bronchodilators or bronchodilators with inhaled corticosteroids (ICS) for the management of stable COPD [1]. Long-acting bronchodilators have been shown to provide consistent improvement in FEV1 (Forced Expiratory Volume in the first second) [2-4]. Treatment with the ICS/long-acting ß2-agonist combination of fluticasone propionate/salmeterol (FSC) at a dose of 250/50mcg twice-daily via DISKUS (FSC DISKUS) has been shown to result in greater improvement in FEV1 than corresponding doses of fluticasone propionate and salmeterol alone [3]. FSC DISKUS has also been shown to improve exercise endurance time, lung hyperinflation, and reduce exacerbations in patients with COPD [3, 5].
FSC is also available in inhalation aerosol formulations. The inhalation aerosol formulation of FSC is delivered via metered-dose-inhaler and is available in the following 3 strengths (doses) expressed as drug delivered from the actuator: 45/21mcg (90mcg/42mcg twice-daily), 115/21mcg (230/42mcg twice-daily), and 230/21mcg (460/42mcg twice-daily). FSC in the MDI is formulated with the hydrofluoroalkane 134a (HFA) propellant which was developed as a non-ozone depleting alternative to the chlorofluorocarbon MDI. All doses of FSC in the MDI are indicated for the treatment of asthma in the United States (US). The dose of MDI evaluated in this study was 115/21mcg two puffs twice-daily (FSC MDI) which is equivalent to the 250/50mcg dose of FSC DISKUS that is indicated for the treatment of COPD in the US. While the FSC DISKUS device is widely used for the treatment of COPD, there may be instances when the MDI is preferred by the patient or when the DISKUS is not a clinical option. The MDI device may be the only option for patients with COPD that have a treacheostomy or who are intubated. Additionally, FSC DISKUS contains lactose and is contraindicated in patients with significant allergic reactions to lactose or milk protein. The purpose of this study was to provide comparative information on the efficacy and safety of the FSC MDI and FSC DISKUS in patients with COPD.
MATERIALS and METHODOLOGY
Patients
Inclusion Criteria: a) diagnosis of COPD; b) current or former smokers with at least a 10 pack year history; c) aged > 40 years; d) post-bronchodilator FEV1 of > 0.70L and ≤ 70% predicted normal (or if FEV1 ≤ 0.70 L, then ≥40% of predicted normal value), and a post-albuterol FEV1/FVC ratio of ≤ 0.70.
Exclusion Criteria: a) current diagnosis of asthma; b) respiratory diagnosis considered to be significant that was not related to COPD; c) a clinically significant and uncontrolled medical disorder; d) experienced a COPD exacerbation/infection that required corticosteroids and/or antibiotics that did not resolve within 30 days of visit 1; e) a COPD exacerbation that resulted in hospitalization and did not resolve within 3 months of screening; f) abnormal and clinically significant 12-lead electrocardiogram (ECG) at screening ; g) a body mass index (BMI) cut-off > 40kg/m2; h) use of nocturnal positive pressure such as continuous positive airway pressure or bi-level positive airway pressure was exclusionary. For the purposes of this study, an abnormal ECG was defined as a 12-lead tracing which was interpreted as (but not limited to) myocardial ischemia, clinically significant arrhythmias or left bundle branch block.
Study Design: This was a randomized, double-blind, double-dummy, parallel group study that was conducted at 16 research sites in the United States. Each site received institutional review board or ethics committee approval for the study and all patients provided written informed consent. The patients completed an 8 -14 day run-in period on only short- acting ß2 agonists. Following run-in patients were randomized to FSC MDI twice-daily or FSC DISKUS twice-daily for a period of 3 months. Study visits were conducted at screening, day 1 (randomization), and after 4, 8, and 12 weeks of treatment. Patients were stratified based on FEV1 response to albuterol (400µg) at screening to provide a similar distribution of albuterol-responsive and non-responsive patients in each group. Albuterol-responsive was defined as an increase in FEV1 of ≥ 200 mL and ≥ 12% from baseline. The use of concurrent inhaled long-acting bronchodilators (ß2-agonist and anticholinergic), ipratropium/albuterol combination products, oral ß2-agonist, inhaled corticosteroids, and theophylline preparations were not allowed during the run-in and double-blind treatment periods.
Measurements: The primary endpoint was 2 hour post-dose FEV1 at endpoint. Endpoint was defined as the last 2-hour post-dose FEV1 measurement obtained during the 12-week treatment period. Baseline was defined as the pre-dose FEV1 measure from Visit 2 (randomization). Secondary efficacy measures were AM (morning) pre-dose FEV1 and AM peak expiratory flow (PEF) over weeks 1-12. Other efficacy measures were 2 hour post-dose Forced Vital Capacity (FVC), AM pre-dose FVC at endpoint and supplemental albuterol use over weeks 1-12. AM PEF measurement and supplemental albuterol use was recorded by the patient daily on diary cards. Quality of life measures were not assessed in this study.
Safety was evaluated by recording all adverse events (AE’s). Adverse events were recorded by patients throughout the study on daily diary cards. Adverse events were collected in eCRFs at all clinic visits and during a final follow-up phone call.
A COPD exacerbation was defined as worsening of dyspnea, cough, and/or sputum beyond day-to-day variability requiring treatment with systemic (oral or parenteral); corticosteroids and/or antibiotics, and/or requiring hospitalization.
Statistical Analysis: The sample size for this study was calculated to assess non-inferiority of the FSC MDI treatment response to the FSC DISKUS treatment response for the primary efficacy endpoint of 2-hour post-dose FEV1. Using 75mL as the non-inferiority criterion bound (lower confidence limit of -75mL) and a standard deviation estimate of 185mL, it was estimated that a sample size of 125 patients per treatment group would provide approximately 90% power to assess non-inferiority of FSC MDI to FSC DISKUS based on a one-sided significance level of 2.5%.
The primary analysis population was the Intent-to-Treat (ITT) population. The ITT population included all patients who had been randomized to study drug. The per protocol population included all patients in the ITT population with no protocol deviations that may have impacted treatment response. As this study was designed to show non-inferiority, analysis of the per protocol population was of importance and served as a confirmatory analysis for the primary analysis of the ITT population.
The primary efficacy analysis was mean change from baseline in 2-hour post-dose FEV1 compared between the two treatment groups at endpoint. An ANCOVA model with terms for treatment group, investigator, reversibility stratum and baseline was used to assess non-inferiority of the primary endpoint. ANCOVA models were also used to compare mean change from baseline values for the secondary and other efficacy measures. AM PEF measures and supplemental albuterol use were compared for the overall 12-week treatment period and spirometry measures were compared at endpoint. Baseline for the diary measures was determined from the 7-day period immediately prior to randomization (Visit 2) and baseline for the spirometry measures was defined as the pre-dose measure from randomization (Visit 2). Treatment group differences from the ANCOVA models were presented as least squares means and standard errors with the corresponding 95% confidence intervals. Statistical programming was performed in a UNIX environment using SAS® Version 9.1.
RESULTS
A total of 365 patients were screened of which 247 were randomized (121 to FSC MDI and 126 to FSC DISKUS). Two hundred and nine (85%) of patients completed the study (106 in FSC MDI and 103 in FSC DISKUS) (Fig. 1). In the FSC MDI group 15 (12%) of the patients were withdrawn while in the FSC DISKUS group 23 (18%) were withdrawn (Fig. 1).
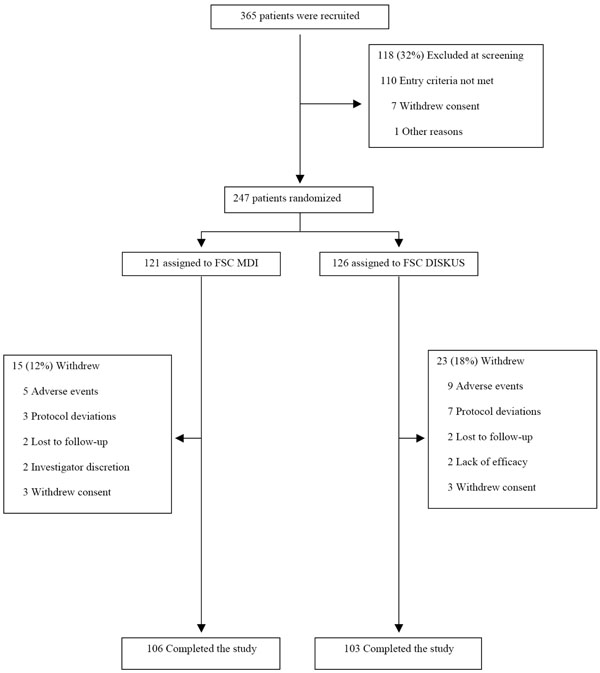
Enrollment of patients and reasons for withdrawal.
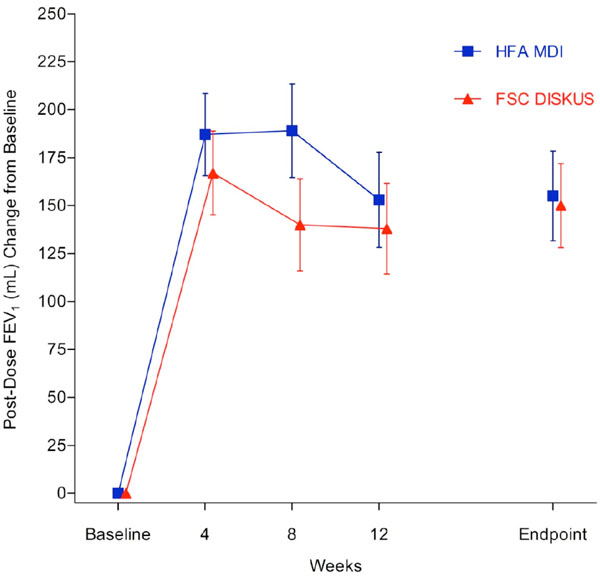
Mean change from baseline in 2-Hour post-dose FEV1 (primary endpoint).
Adverse Events accounted for the main reason for withdrawal in both treatments FSC MDI (n=5) and FSC DISKUS (n=9). Demographics and clinical characteristics are provided in Table 1. Fifty three percent of the patients were male, with a mean age of 63 years (range 40-86 years) and a mean of 58 pack years (range 10-189 years) smoking. Mean (± SD) post bronchodilator FEV1 % predicted was 49.3% (12.3) and 48.4% (11.0) in the FSC MDI and FSC DISKUS groups, respectively.
Demography and Baseline Clinical Characteristics*
| FSC MDI (N =121) |
FSC DISKUS (N = 126) |
|
|---|---|---|
| Age, yrs | 61.6 (40-84) | 63.4 (45-86) |
| Male, n (%) | 66 (55) | 66 (52) |
| Body Mass Index, kg/m2 | 27.3 (16-39) | 26.9 (15-40) |
| Current Smoker, n (%) | 74 (61) | 78 (62) |
| Pack Years- Smoking | 56.2 (14-172) | 60.0 (10-189) |
| Race, n (%) | ||
| White | 110 (91) | 117 (93) |
| African American | 10 (8) | 9 (7) |
| Other | 1 (<1) | 0 |
| Lung Function, Mean (± SD)† | ||
| FEV1 (L) | 1.47 (0.50) | 1.39 (0.40) |
| FEV1 % Predicted | 49.3 (12.3) | 48.4 (11.0) |
| FEV1/FVC | 50.5 (10.0) | 50.3 (10.3) |
* Results represent means (Range) unless otherwise indicated.
† Post-bronchodilator
Supplemental Albuterol Use and AM Peak Expiratory Flow at Baseline and Weeks 1-12
| FSC MDI (N =121) |
FSC DISKUS (N = 126) |
|
|---|---|---|
| Albuterol Use, (puffs/day) Mean (SE) | ||
| Baseline | 4.4 (0.38) | 4.4 (0.34) |
| Weeks 1-12 | 2.4 (0.29) | 2.3 (0.27) |
| LSM difference | 0.1 (0.28) | |
| AM Peak expiratory flow, L/min Mean (SE) | ||
| Baseline | 200.6 (7.14) | 201.0 (6.20) |
| Weeks (1-12) (1-12) | 222.7 (8.10) | 218.0 (6.85) |
| LSM difference | 2.3 (3.55) | |
Summary of Adverse Events
| FSC MDI (N =121) | FSC DISKUS (N = 126) | |
|---|---|---|
| AE’s occurring in >2% of patients in any group during treatment, n (%) | 54 (45) | 59 (47) |
| Any event | 54 (45) | 59 (47) |
| Headache | 10 (8) | 8 (6) |
| Nasopharyngtis | 5 (4) | 8 (6) |
| Cough | 4 (3) | 5 (4) |
| Sinusitis | 3 (2) | 6 (5) |
| Oropharyngeal pain | 5 (4) | 2 (2) |
| Other AE’s of interest, n (%) | ||
| Pneumonia | 0 | 2 (2) |
| Eye disorders† | 0 | 0 |
| Bone disorders†† | 1 (<1) | 1 (<1) |
| Death | 0 | 0 |
† Cataracts and glaucoma.
†† Foot fracture.
Primary Endpoint (2 Hour Post-Dose FEV1): The 2 hour post-dose FEV1 at endpoint met the a priori definition of non-inferiority (-2mL LS mean difference, (-64, 59ml) 95% confidence interval and non-inferiority p = 0.021). The mean (SE) 2 hour post-dose FEV1 for the FSC MDI group was 1289 (44) mL at baseline and 1446 (49.2) mL at endpoint. For the FSC DISKUS group the 2 hour post-dose FEV1 mean (SE) was 1228 (38.6) mL at baseline and 1372 (43.8) mL at endpoint. The mean (SE) 2 hour post-dose FEV1 change from baseline for the FSC MDI group was 155 (23.4) mL and 150 (21.9) mL for the FSC DISKUS arm (Fig. 2).
Secondary and Other Efficacy Measures: The AM pre-dose mean FEV1 (SE) for the FSC MDI was 1365 mL (47.3) at endpoint. For the FSC DISKUS group the mean (SE) was 1299 (42.9) mL at endpoint. The least squares mean (SE) difference was -8 (28.9). The 2 hour post-dose FVC mean (SE) for the FSC MDI was 2630 (74.2) mL at baseline and 2825 (73.5) mL at endpoint. For the FSC DISKUS group the mean (SE) was 2486 (59.2) mL at baseline and 2720 (67.4) mL at endpoint. The least squares mean (SE) difference was -33 (49.9) mL. The AM pre-dose FVC mean (SE) for the FSC MDI was 2675 (72.1) mL at endpoint. For the FSC DISKUS group the mean (SE) was 2618 (66.3) mL at endpoint. The least squares mean (SE) difference was -74 (48.1) mL. The least squares mean (SE) difference at weeks 1-12 for supplemental albuterol use between the groups was 0.1 (0.28) puffs per day (Table 2). The least squares mean (SE) difference at weeks 1-12 for AM PEF between the groups was 2.3 (3.55) L/min (Table 2). Improvements in the secondary and other efficacy measures were similar for both the FSC DISKUS and FSC MDI groups. Differences between the treatment groups were not statistically significant.
Safety: A total of 5 (4%) and 9 (7%) patients were withdrawn from the study as a result of AE’s in the FSC MDI and FSC DISKUS groups, respectively. The most common AE leading to withdrawal from the study in the FSC MDI group was worsening of COPD 3 (2%). In the FSC DISKUS group worsening of COPD 2 (2%), pneumonia 2 (2%) and bronchitis 3 (2%) were the most common AEs leading to withdrawal from the study. All other AE’s leading to withdrawal occurred <1%. In the FSC MDI 45%, and 47% in the FSC DISKUS group reported any AE during treatment (Table 3). Candidiasis was reported by 1 (<1%) patient in the FSC MDI treatment group and by 3 (2%) patients in the FSC DISKUS treatment group. COPD exacerbations were reported for 10 (8%) patients in the FSC MDI group and 16 (13%) patients in the FSC DISKUS group. The frequency of AE’s which occurred in > 2% of patients in any group during treatment is provided in Table 3.
DISCUSSION
Although previous randomized clinical trials (RCT) have demonstrated the safety and efficacy of the FSC DISKUS in COPD patients [3, 5-8], this is the first RCT to evaluate the safety and efficacy of FSC in inhalation aerosol formulation delivered via MDI in patients with COPD. Other studies [9-11] have explored the efficacy and safety of these different formulations in asthma and were able to demonstrate that efficacy and safety were comparable.
Similarly, we found that FSC delivered via an MDI was as effective (non-inferiority) as when delivered via a dry powder in the DISKUS based on the primary endpoint of 2-hour post-dose FEV1. Other measures of lung function (FVC and PEF both pre-dose and post-dose) were also similar, suggesting that the different formulations have similar efficacy when used in a clinical trial setting.
With regards to safety, the incidence of adverse events, exacerbations, and pneumonia were similar for the two formulations. There were very few events in the study overall, a not unexpected finding given the small study size and the length of the trial. As in many COPD trials, headache and upper respiratory complaints were reported most commonly. Topical steroid adverse events were infrequent and did not occur at a frequency >2% in our trial, again not surprising in a trial of this size where patients were instructed on how to use both devices at the start of the trial. However, this trial was not designed to detect small differences in adverse events, so no definitive conclusions can be drawn from this data. Finally, no spacer devices were used in this trial and this may also have an effect on topical steroid side effects such as thrush and dysphonia.
Demonstrating efficacy can be influenced by compliance as well as handling of the drug delivery device. Several studies have compared the compliance and handling of the DISKUS and the MDI [12-15]. Khassawneh et al. in a prospective observational analysis evaluated handling of inhaler devices in pulmonary clinics and reported that the DISKUS had the lowest rate of incorrect handling while the MDI had the highest rate of incorrect handling [13]. We did not measure handling of MDI or Diskus, but the patients in our study did receive instructions on how to use their devices at each visit.
There are several limitations of this study. The primary limitation was the size and duration of the trial. This first study of the MDI formulation in COPD was designed to look for differences in effects on lung function between the active ingredients (fluticasone propionate and salmeterol) delivered via a dry powder formulation in the DISKUS and a liquid formulation in the MDI. A larger trial of longer duration may have demonstrated differences in other efficacy endpoints beyond lung function (e.g. exacerbations, dyspnea and quality of life) and in adverse events and safety endpoints. In addition, in this clinical trial setting, patients were instructed on how to use their devices at each study visit. This is unlikely to occur in a typical practice setting, so ease of use of the device and its impact on patient compliance which may affect efficacy must also be taken into account.
Finally, there has been speculation that particle size emitted from the device may affect the efficacy of a drug. In this study, we were unable to see any differences in the efficacy due to the different formulations. While the distribution of particle size emitted from the inhalers may be slightly different, that did not appear to impact either the trough lung function (pre-dose) or the maximum lung function (2-hour post dose when salmeterol is at its maximum).
While the MDI formulation is not indicated for the treatment of COPD in the United States, it is important to provide clinical information on its safety and efficacy in patients with COPD. There may be instances in which COPD patients are unable to use the DISKUS or have a strong preference for a MDI device. The MDI device may be the only option for patients with COPD that have a treacheostomy or who are intubated. In addition, FSC DISKUS contains lactose and is contraindicated in patients with significant allergic reactions to lactose or milk protein. In these patients the MDI may be an option for the clinician to consider.
CONCLUSION
This study demonstrated that the Fluticasone Propionate/Salmeterol Hydrofluoroalkane 134a Metered-Dose-Inhaler 230/42mcg showed similar efficacy when compared with the Fluticasone Propionate/Salmeterol DISKUS 250/50mcg in patients with COPD. Additionally, this study showed the MDI and DISKUS formulations had similar adverse event profiles.
CONFLICT OF INTERESTS
Andras Koser has received COPD related research funding from GlaxoSmithKline.
Jan Westerman has received COPD related research funding from GlaxoSmithKline.
Sanjay Sharma, Amanda Emmett and Glenn Crater are GlaxoSmithKline employees and own stock in the company.
TRIAL REGISTRATION
GlaxoSmithKline Study ADC111117, NCT00633217.
ACKNOWLEDGEMENTS
Funding for this study was provided by GlaxoSmithKline (NCT00633217). All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors. The authors wish to acknowledge Andrea Morris, employed by GSK, for editorial assistance and critical review during the development of this manuscript.

