All published articles of this journal are available on ScienceDirect.

Inspiratory Flows and Volumes in Subjects with Cystic Fibrosis Using a New Dry Powder Inhaler Device
Authors Info & Affiliations
Abstract
Introduction:
Drug inhalation via a dry-powder inhaler (DPI) is a convenient, time efficient alternative to nebulizers in the treatment of cystic fibrosis (CF). Efficient drug administration via DPIs depends on the device resistance and adequate (≥ 45L/min) inspiratory flows and volumes generated by individuals. Dry-powder mannitol is delivered using a RS01 breath-actuated device developed by Plastiape, for Pharmaxis. The study aim was to determine in vivo if CF patients’ inspiratory flows and volumes are adequate to use the RS01 DPI device.
Materials and Methodology:
An open, non-interventional study; enrolled 25 CF subjects, aged ≥ 6 years with FEV1 ≥ 30 to < 90‰ predicted. Inspiratory flows and volumes were measured when subjects inhaled in a controlled manner through the RS01 device in series with a spirometer.
Results:
The mean inspiratory volume (IV) of CF subjects was 1.83L ± 0.97. Their achieved mean ± SD peak inspiratory flow (PIF) was 75.5 ± 27.2L/min. Twenty-three subjects (92%) achieved PIF of > 45L/min with the inhaler device; eighteen of those subjects (78%) had a baseline FEV1 of > 1L.
Conclusion:
Use of the RS01 DPI device allowed adequate inspiratory flow and volume for dispersion of dry-powder mannitol in CF patients.
1.. INTRODUCTION
Inhalation of dry powder formulations via a dry-powder inhaler (DPI) is a convenient and time-efficient alternative to inhalation of wet aerosols via nebulizers in the treatment of respiratory diseases such as cystic fibrosis (CF) or bronchiectasis. Generally, adequate administration of drugs via DPIs depends on the resistance of the device and adequate inspiratory flows and volumes generated by individuals. These may vary according to the device being used for a particular formulation and the patient’s lung function impairment.
Encapsulated precision spray-dried mannitol for inhalation is used in the currently registered diagnostic product Aridol®/Osmohale® to identify bronchial hyper-responsiveness as well as in the CF therapeutic Bronchitol®. It is also being used in clinical trials as a potential treatment in subjects with non-CF bronchiectasis. Each Bronchitol® capsule (gelatin size 3) contains 40 mg of spray-dried mannitol powder with no excipients. Spray-dried mannitol powder consists of particles that are of respirable size [1] (< 5 µm in diameter) in sufficient quantity and spherical in shape to allow them to disperse well in an aerosol. The mannitol dose, 400 mg (10x40 mg capsules), required to generate a significant osmotic effect in the lung, can be delivered in less than 5 minutes by a simple hand-held DPI, which requires no power source and provides a convenient and hygienic delivery system.
A convenient and efficient breath-actuated portable device, the dry powder inhaler type RS01 Model 7, manufactured by Plastiape S.p.A. Via 1 Maggio 8, 23875 Osnago-Lecco in Italy (CE marked) (Fig. 1A-D), has been approved for use with dry-powder mannitol. Two versions of the RS01 device are available and have been approved for use with two dry-powder mannitol products for inhalation, Aridol® and Bronchitol®; the low resistance (LRes) inhaler with mean resistance 0.021 kPa1/2/L min used in the mannitol bronchial challenge test (Aridol®) and some earlier bronchiectasis studies, and a high resistance (HRes) inhaler with mean resistance of 0.036 kPa1/2/L min which has been trialled and marketed in CF patients and is now being trialled in studies with non-CF bronchiectasis patients [Pharmaxis data on file]. The only difference between the two devices relates to the diameter of the air inlet ports on each side of the inhaler. The HRes has a flange that restricts the flow, resulting in a slightly higher resistance (Fig. 1D).
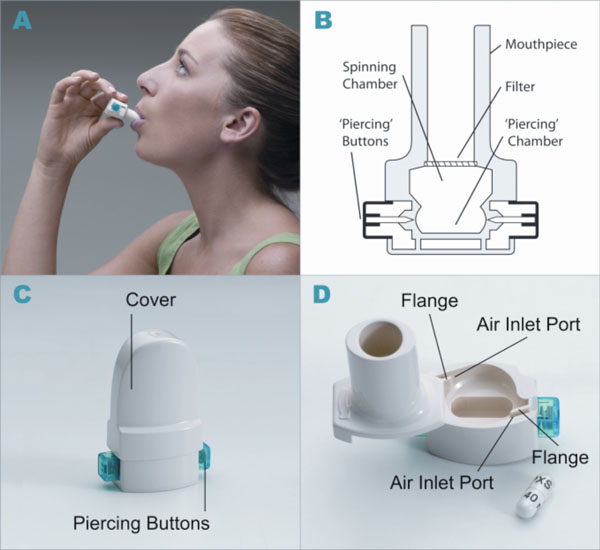
Dry powder inhaler type RS01 model 7. (A) How to use the inhaler device. (B) Representation of the inhaler device. (C) HRes Inhaler with cover and showing piercing buttons. (D) HRes Inhaler showing air inlets and cavity for capsule.
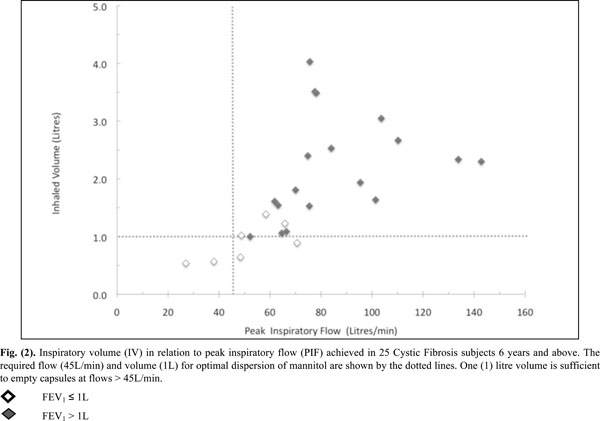
Inspiratory volume (IV) in relation to peak inspiratory flow (PIF) achieved in 25 Cystic Fibrosis subjects 6 years and above. The required flow (45L/min) and volume (1L) for optimal dispersion of mannitol are shown by the dotted lines. One (1) litre volume is sufficient to empty capsules at flows > 45L/min.
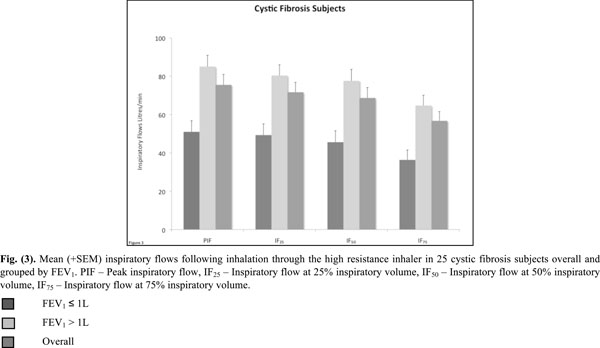
Mean (+SEM) inspiratory flows following inhalation through the high resistance inhaler in 25 cystic fibrosis subjects overall and grouped by FEV1. PIF - Peak inspiratory flow, IF25 - Inspiratory flow at 25% inspiratory volume, IF50 - Inspiratory flow at 50% inspiratory volume, IF75 - Inspiratory flow at 75% inspiratory volume.
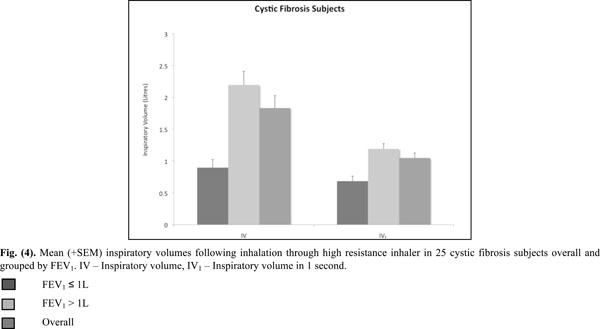
Mean (+SEM) inspiratory volumes following inhalation through high resistance inhaler in 25 cystic fibrosis subjects overall and grouped by FEV1. IV - Inspiratory volume, IV1 - Inspiratory volume in 1 second.
The choice to study and market the HRes device with Bronchitol when it is used therapeutically was made based on patient feedback in clinical studies, which indicated that the HRes provided better ease of use and tolerability than the LRes device in CF and non-CF bronchiectasis.
Depending on age and the degree of lung function impairment, CF patients may not be able to generate optimal flows or volumes for drug delivery through the HRes RS01 DPI. We conducted an open observational non-drug study to confirm the usability of the new dry-powder inhaler RS01 in children and adult subjects with CF, all of whom had inspiratory flow and volumes in the range eligible to use the product [Pharmaxis data on file].
2.. STUDY OBJECTIVES
The primary objective of the study was to determine the inspiratory flow of subjects with CF using the HRes RS01 DPI device.
The secondary objective was to determine inspiratory volumes in CF subjects using the HRes RS01 DPI device.
3.. MATERIALS AND METHODS
3.1.. Recruitment
Subjects were recruited from within two Australian CF treatment centres. All patients provided written informed consent.
3.2.. Inclusion / Exclusion Criteria
The subjects eligible to participate were:
Subjects with a confirmed diagnosis of CF by sweat test or genotype, aged at least six years, and with FEV1 ≥ 30% and < 90% of predicted [2,3].
Exclusion criteria included subjects who had uncontrolled asthma or previously demonstrated airway hyperresponsiveness to mannitol, or were considered “terminally ill” or listed for lung transplantation, lung transplant recipient, significant episode of haemoptysis (>60 ml) in the three months prior to study enrolment, myocardial infarction, cerebral vascular accident, major ocular, abdominal, chest or brain surgery in the three months prior to enrolment. Also, known cerebral, aortic or abdominal aneurysm, breast feeding or pregnant subjects or using beta blockers or had any other condition which in the investigator’s opinion may put the subject at significant risk, confound results or interfere significantly with the patient’s participation in the study were excluded.
3.3.. Sample Size
Due to the exploratory nature of this study no formal sample size calculations were performed.
3.4.. Study Design
A Phase IV, observational, open label, non-interventional, multi-centre, single visit clinical study primarily measuring the inspiratory flow through the HRes RS01 inhaler device in a group of subjects with CF. (ClinicalTrials.gov Number NCT00779077)
At baseline each potential subject was examined to determine eligibility for the study. This included written informed consent, collection of their respiratory history and demographic information and measurement of FEV1 % predicted.
3.5.. Setting
Data were collected in two tertiary hospitals in Australia.
3.6.. Outcomes
The following measures were collected: PIF, IF25, IF50, IF75, IV and IV1.
3.7.. Study Protocol
A standard spirometer (calibration checked daily) was used to measure the forced expiratory volume in one second (FEV1) according to ATS/ERS criteria [4]. Spirometers used at each centre were: RCH – Micro Medical™ Microlab Mk8 Spirometer Cat. ML3500 (CareFusion) and RPAH – Micro Medical SpiroUSB™ Cat. ML2525 (CareFusion). The same spirometer was used for all subjects at each site.
Spirometry was conducted with the subjects seated in a comfortable position. Subjects were given an opportunity to practice the technique inspiring comfortably at 60L/min. The HRes RS01 DPI device, containing an empty but unpierced capsule, was placed in series with the spirometer, allowing the inspiratory flows and volumes to be recorded on a computer. In this study an empty capsule was employed in lieu of a 40 mg capsule of Bronchitol. An empty capsule is similar to the empty device and dummy device methods employed by other researchers when measuring flows through new devices. The empty unpierced capsule was changed between each inspiration. The subjects were instructed to exhale away from the device then place the mouthpiece in their mouth and create a firm seal with their lips. The subjects took 3 to 5 steady deep controlled inspirations using the HRes inhaler and the inspiratory flow and volume were measured during each inspiration. Inspiratory flow measures including PIF, IF25, IF50, and IF75 were calculated using three out of five inhalations following the study procedure. The mean of the three trials having the greatest PIF of the five trials performed was used for the following inspiratory measures: PIF, IF25, IF50, IF75, IV and IV1.
3.8.. Safety
During this non-interventional study, all subjects underwent the same testing which involved an inspiratory manoeuvre (three to five deep, controlled inspirations) via a spirometer to which the DPI device containing empty, unpierced capsules was placed in series. The measurements were made at one visit only and the subjects were not administered a drug or any intervention. All adverse events which occurred during the inhalations and up to 15 minutes after completion were recorded by the investigator.
3.9.. Statistical analyses
Descriptive statistics of continuous measures consist of the number of observations (n), mean, standard deviation (SD), median, minimum and maximum values and 95% confidence intervals. Categorical variables are described using counts of frequency and percentage of group (n(%)).
Peak inspiratory flow (PIF) and maximal inspiratory flows at 25%, 50% and 75% of maximal inspiratory volume (IF25, IF50 and IF75) were summarised and tabulated overall and by baseline FEV1 groups; ≤ 1L and > 1L. A two independent sample t-test was used to determine whether differences existed between FEV1 groups for each measure.
3.10.. Ethics
The study was approved at both study sites; the Royal Children’s Hospital Research Ethics Committee, Melbourne, Australia and the Sydney South West Area Health Service, Ethics Research Committee in Sydney, Australia, prior to commencement of the study. Subjects provided written informed consent for their participation in the study.
4.. RESULTS
4.1.. Patient Demographics and Disposition
Twenty-five subjects diagnosed with CF were enrolled in the study (Table 1). All subjects completed the study. Seven of the subjects (28%) had a baseline FEV1 ≤ 1L and 18 subjects had a baseline FEV1 > 1L (Table 1).
Baseline Characteristics (All Subjects)
| Parameter | Cystic Fibrosis | ||
|---|---|---|---|
| Overall (n=25) |
FEV1 ≤ 1L (n=7) |
FEV1 > 1L (n=18) |
|
| Age, years: Mean (SD) [Range] Gender: Male Female |
16.0 (8.8) [6 – 37] 10 (40%) 15 (60%) |
9.4 (3.3) [6 – 15] 2 (29%) 5 (71%) |
18.6 (9.0) [6 – 37] 8 (44%) 10 (56%) |
| FEV1 (L) Mean (SD) [Range] Median |
1.53 (0.74) [0.40 – 3.23] 1.36 |
0.79 (0.23) [0.40 – 1.00] 0.91 |
1.82 (0.65) [1.02 – 3.23] 1.78 |
| FEV1 (% predicted) Mean (SD) [Range] Median |
57.6 (18.8) [30 – 89] 57 |
47.7 (20.3) [30 – 82] 42 |
61.4 (17.3) [33 – 89] 65 |
Inspiratory Flows and Volumes in Cystic Fibrosis Subjects Overall and Grouped by FEV1
| Group | Mean Parameters (SD) | |||||
|---|---|---|---|---|---|---|
| Baseline FEV1 | PIF (L/min) |
IF25 (L/min) |
IF50 (L/min) |
IF75 (L/min) |
IV (L) |
IV1 (L) |
| n=25 Overall (SD) Range |
75.5 (27.2) 27.2 – 142.8 |
71.7 (25.7) 25.8 – 130.2 |
68.7 (27.1) 22.2 – 136.2 |
57.9 (24.1) 21.0 – 122.4 |
1.83 (0.97) 0.54 – 4.03 |
1.05 (0.39) 0.39 – 1.96 |
| n=7 FEV1 ≤ 1L (SD) Range |
51.0 (15.4) 27 – 70.8 |
49.3 (15.4) 25.8 – 68.4 |
45.6 (15.7) 22.2 – 66.6 |
36.4 (13.7) 20.4 – 54.6 |
0.90 (0.33) 0.54 – 1.39 |
0.68 (0.21) 0.39 – 0.92 |
| n=18 FEV1 > 1L (SD) Range |
85.1 (24.9) 52.2 – 142.8 |
80.4 (23.8) 48.0 – 130.2 |
77.6 (25.4) 46.8 – 136.2 |
64.7 (22.7) 25.8 – 120.6 |
2.20 (0.89) 1.00 – 4.03 |
1.19 (0.35) 0.76 – 1.96 |
PIF - Peak inspiratory flow, IF25 - Inspiratory flow at 25% inspiratory volume, IF50 - Inspiratory flow at 50% inspiratory volume, IF75 - Inspiratory flow at 75% inspiratory volume, IV - Inspiratory volume, IV1 - Inspiratory volume in 1 second.
4.2.. Lung Function
All inspiratory flows and volumes, overall and grouped for FEV1 ≤ or > 1L, are shown in Table 2 and illustrated in Figs. (2-4). Overall, 23 patients (92%) generated a PIF of > 45L/min. The two patients unable to generate this produced a PIF of 27 and 38L/min and had a baseline FEV1 of 0.57 (42% predicted) and 0.71 (35% predicted). The lowest PIF of 27.0L/min was measured in a 7 year old female, who also had asthma.
Patients (n=18) with a baseline FEV1 > 1L achieved a PIF of > 45L/min and had an IV of > 1L. Out of those with a baseline FEV1 ≤ 1L (n=7), five generated a PIF > 45L/min with 3 having an IV of > 1L.
The lowest IF was an IF75 of 21L/min in the same 7 year old subject mentioned previously. Only 4 subjects had IV values < 1.0L (Fig. 2) and were amongst the group who had a baseline FEV1 ≤ 1.0L.
DISCUSSION
The findings support the use of the HRes RS01 device for delivering dry powder mannitol to the lower airways in subjects with cystic fibrosis.
The important determinants for optimal dispersion of drugs via DPI are the device resistance and the inhalation flow and volume of the subjects [5-10]. The resistance of the RS01 HRes device of 0.036 kPa1/2/L min is within the range of the resistance of DPIs used for delivering other dry powder formulations to the lung [5,6]. The flows that the patients generate inhaling through the device are of critical importance as they provide the energy for optimal dispersion of the powder [6]. However the flows patients generate depend on the resistance of the device and on their own lung function. In-house in vitro data comparing the HRes inhaler with Bronchitol at 45 and 60L/min showed that the fine particle dose (FPD) was the same at both flow rates. The decision to use the HRes device for the phase III CF trials was made following patient feedback on ease of use and tolerability of the HRes device, making it the most suitable for frequent administration of inhaled mannitol over the long-term.
In vitro testing of the device for delivered dose uniformity (DDU) and aerodynamic particle size distribution (APSD) of dry powder mannitol, in accordance with the European Pharmacopoeia (Ph.Eur.) [11] and United States Pharmacopeia (USP) [12], showed that the HRes device gave comparable delivered dose and APSD between 45 and 100L/min. The Fine Particle Dose (<5 µm) test results were all within a range of ±6% and the MMAD (mass median aerodynamic diameter) results were all within a range of 3.9 to 4.1 µm when using the standard test volume of 4L. The mean delivered dose test results were all within a range of ±13% when using test volumes of 1L to 4L [Pharmaxis data on file]. Lower flow rates and volumes tested in vitro resulted in lower delivered doses. The delivered dose was reduced by approximately 20% at 28L/min when using an inhalation volume of 2L. When the inhalation volume was reduced to 1L at 28L/min the reduction in the delivered dose was approximately 35%.
The majority of the CF subjects (92%) using the HRes device generated a PIF of 45L/min. Only two subjects (8%) did not generate this flow. Patients in the CF group with FEV1 > 1L generated higher results for all parameters, however 71% of patients with baseline FEV1 ≤ 1L were still able to achieve PIF rates > 45L/min. The resistance of the HRes RS01 device is similar to the one used by Tiddens et al. [5] (0.038 kPa1/2/L min) in a study with 96 CF subjects. Using this inhaler, Tiddens et al. [5] found that a PIF of 45L/min was achieved by 100% of subjects >18 years, 96% between 11-18 years and 88% between 6-10 years of age. These results are in good agreement with the results of the present study.
This study confirms that the minimum PIF of 45L/min is able to be generated by the majority of patients, the exception being young patients with impaired lung function where a second inhalation may be necessary [13]. In this study subjects practiced taking a deep and steady inhalation at 60L/min. We were simply interested in the flows that subjects generated without maximum efforts. The rate of rise of inhalation flow was not measured and its effect on the deaggregation of mannitol for optimal MMAD is not known. It has been shown though that the inhalation flow profile is very reproducible within the same individual but highly variable between individuals [14]. While the rate of rise of inhalation flow did not correlate with the deposited mannitol in the lung, the mannitol deposited in the lung did correlate with the peak inspiratory flow [14]. Based on the results of the IF25, IF50 and IF75, it is obvious that the highest IF was achieved in the first 50% of the IV (Table 2).
Sufficient inspired volume is necessary for emptying the capsule. The majority of CF (84%) subjects had inspired volume greater than 1L. It is recommended that in clinical practice patients should check to see that the capsule has emptied and if not then the inhalation should be repeated until the capsule appears empty. A second inhalation is recommended by the ERS/ISAM Task force when the powder is in capsules, as is the case with mannitol [15].
In two large phase III clinical studies in CF [16,17] patients were instructed to use the inhaler and inhale the mannitol in a similar way to that described for this current research and the ERS/Task force guidelines [15]. In the two large phase III clinical studies [16,17], patients over the age of 6 years, with FEV1 ranging between 29.2% and 96.1% predicted (0.58L and 4.73L), were treated with 400 mg of inhaled mannitol twice daily and had clinically significant sustained improvement in their FEV1, indicating that mannitol was delivered in an effective dose.
CONCLUSION
Together with in vitro laboratory testing for dose uniformity and aerodynamic particle size distribution for dry powder mannitol, and clinical results from phase III studies, this study confirms that the majority of subjects with CF were able to generate the inspiratory flows and volumes necessary to successfully operate the RS01 DPI designed for the inhalation of mannitol.
Trial registration: ClinicalTrials.gov Identifier: NCT00779077.
ABBREVIATIONS
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest.
ACKNOWLEDGEMENTS
Author’s Role
All authors helped to interpret data, write the manuscript and have seen and approved the final version.
Dr. Charlton designed the study, approved the statistical plans and was the Sponsor’s Responsible Medical Officer. He is also identified as the guarantor of the paper, taking responsibility for the integrity of the work as a whole, from inception to published article. Drs Anderson, Elkins and Robinson were the Investigators at respective investigational sites and had full access to all the data in the study and have no conflict to disclose relevant to this manuscript. Their institutions received standard clinical trial support from Pharmaxis. No Investigator received any personal funding to participate in the study. Ms Perry worked on the OSM403 CF study. Dr Daviskas helped with the preparation of the manuscript.
Dr. Anderson, Clare Perry and Dr Daviskas are employed by Sydney South West Area Health Service. All hold shares in Pharmaxis Ltd that they purchased but they do not hold any options.
Dr. Charlton is the Medical Director of and holds stock options in Pharmaxis Ltd.
Funding
Pharmaxis Ltd.
Role of the Funding Source
The study sponsor participated in the study design, data collection, data analysis, data interpretation and writing of the reports. Following completion of the trials, the data were held and analysed by the sponsor. All authors had full access to all data and Mark Elkins had final responsibility for the publication.
We thank Ruth Martens who worked on the flow studies for Pharmaxis and Carina Flodin, Development and Technical Services Co-ordinator at Pharmaxis.

