All published articles of this journal are available on ScienceDirect.

The Three A’s in Asthma – Airway Smooth Muscle, Airway Remodeling & Angiogenesis
Abstract
Asthma affects more than 300 million people worldwide and its prevalence is still rising. Acute asthma attacks are characterized by severe symptoms such as breathlessness, wheezing, tightness of the chest, and coughing, which may lead to hospitalization or death. Besides the acute symptoms, asthma is characterized by persistent airway inflammation and airway wall remodeling. The term airway wall remodeling summarizes the structural changes in the airway wall: epithelial cell shedding, goblet cell hyperplasia, hyperplasia and hypertrophy of the airway smooth muscle (ASM) bundles, basement membrane thickening and increased vascular density. Airway wall remodeling starts early in the pathogenesis of asthma and today it is suggested that remodeling is a prerequisite for other asthma pathologies. The beneficial effect of bronchial thermoplasty in reducing asthma symptoms, together with the increased potential of ASM cells of asthmatics to produce inflammatory and angiogenic factors, indicate that the ASM cell is a major effector cell in the pathology of asthma. In the present review we discuss the ASM cell and its role in airway wall remodeling and angiogenesis.
AIRWAY WALL REMODELING
One of the distinct features of the asthmatic lung is airway wall remodeling, which refers to persistent cellular and structural changes in the airway wall. In progressive disease, airway remodeling includes epithelial goblet cell hypertrophy, enhanced collagen deposition and airway wall hyperplasia. As early as 1922, it was recognized that the lungs of asthmatic patients are characterized by an increased thickness of the airway wall, in particular by an enlarged bulk of smooth muscle mass [1]. We now know that this increase is predominantly caused by mesenchymal (or: airway tissue forming) cells and a correlation between airway wall thickness and disease severity has been established [2-4]. In the asthmatic airway wall, we usually observe basement membrane thickening, hyperplasia and hypertrophy of ASM cells, and increased vascularity. In addition, we often observe epithelial cell shedding and thickening, goblet cell hyperplasia, and mucus gland hypertrophy [5-8]. It remains a matter of debate whether the immune system is the key player in asthma pathogenesis or whether tissue-forming lung cells are sufficient to explain the development of asthma [9]. The latter view is supported, however, by the observation that airway wall remodeling is observed in preschool children without clinical signs of inflammation [10-17]. Thickening of the airway wall sub-epithelial cell layers, together with the edematous effects of increased numbers of blood vessels results is airway narrowing, the major long term complication of asthma [18]. Fig. (1A-D) illustrates several of the pathologies of the remodeled airway wall as it can be observed in asthma patients.
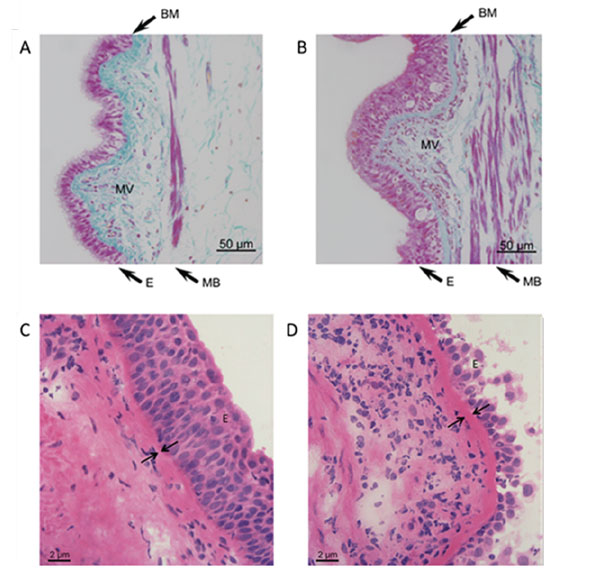
Top: Milligan’s trichrome stained sections of airway tissue from non-asthma (A) and asthma (B) patients. Images are representative of tissues obtained from 3 non-asthmatic and 3 asthmatic patients. Nuclei and muscle: magenta, collagen: green, RBC: orange. Note epithelial hyperplasia, thickening of muscle bundles and basement membrane, and increased microvessel density in asthmatic airways. E = epithelium, MB = muscle bundles, BM = basement membrane, MV = microvessels. Bottom: Light microscopic images of Haematoxylin- Eosin-stained airway tissue sections obtained from a non-asthma (C) and an asthma (D) patient. Note the increased thickening of the basement membrane in the asthmatic airways. E = epithelium, arrows are indicating the basement membrane. (Figure adapted from references 89 and 106).
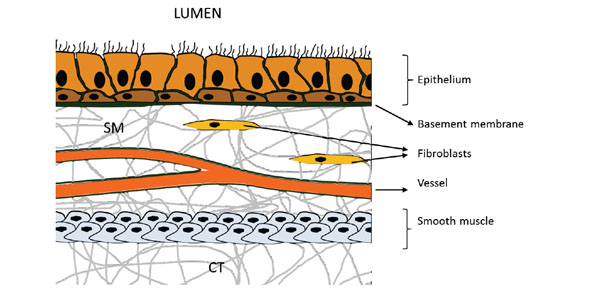
From the airway lumen down towards the connective tissue (CT) in the deeper regions of the airway wall we successively encounter the epithelium, the basement membrane, the submucosa (SM) containing mesenchymal cells (predominantly fibroblasts) and microvessels, and finally smooth muscle cells.
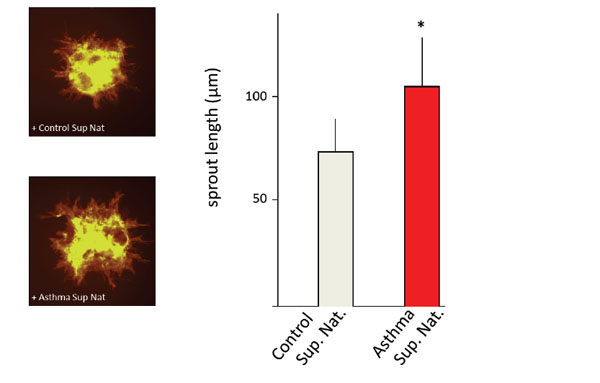
Neovascularization (spout-outgrowth from human vascular endothelial cells (EC)-spheroids is increased in the presence of super natant (Sup Nat) obtained from asthmatic ASM cells (left panel). Images are representative for illustrating sprout outgrowth from ECspheroids incubated with supernatants of non-asthmatic and asthmatic ASM cells (as indicated). Lengths of sprouts are indicated as the calculated mean (±SD) of the longest 10 sprouts. * p < 0.05. Further details about sprouting and angiogenesis assays can be found in references 89 and 106.
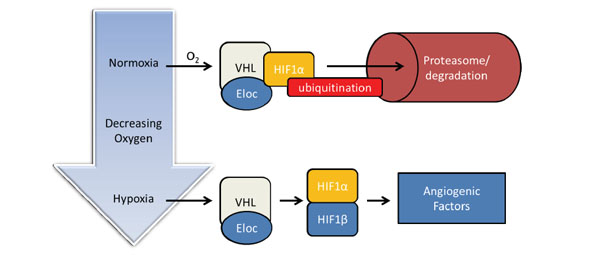
Schematic illustration of the HIF oxygen-sensing pathway. Under normal oxygenation HIF1α is ubiqitinated and degraded in the proteasome. Low oxygen concentrations (hypoxia) induce HIF1β, followed by formation of the hetero-dimeric HIF transcription factor, which is required for the transcription of angiogenic factors (for details see text).
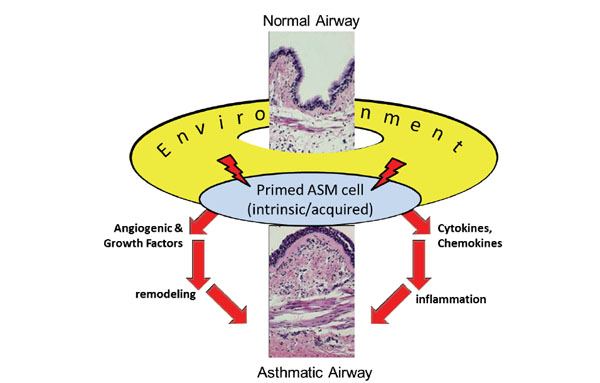
Schematic presentation of the proposed central role of the airway smooth muscle (ASM) cell in airway wall remodeling. Due to predisposition (intrinsic) and/or environmental (acquired) cues (allergens, hypoxia), ASM cells of patients with asthma are “primed” to release abnormal levels of remodeling factors (angiogenic and growthfactors, as well as cytokines and chemokines).
CELLS OF THE AIRWAY WALL
Traveling from the airway lumen down towards the connective tissue in the deeper regions of the airway wall, we first encounter the epithelium (Fig. 2). Epithelial cells have long been considered merely a mechanical barrier protecting the body from the environment. It is the first barrier against all influences from outside (allergens, pollutants, temperature, etc.) and has been reported to be damaged and metaplastic (pseudostratisfied) in asthma. Epithelial cells produce a vast spectrum of cytokines and chemokines [19, 20]. It has been shown that epithelial cell proliferation in asthmatics is impaired, which was attributed to altered level of several markers of proliferation such as proliferating cell nuclear antigen (PCNA), Ki67, and p21 [21-23]. Furthermore, in asthmatics the composition of the extracellular matrix, which forms hemi-desmosomes with the epithelial cells, is altered and the expression of tight-junction proteins is reduced [24-26]. These findings suggest a chronic injury of the airway epithelium with reduced ability to repair and may underlay the increased sensitivity to aspecific (e.g. cold air) and specific stimuli (e.g. smoke, ozone), as well as the increased release of pro-inflammatory factors [23, 27, 28].
Directly beneath the epithelium we encounter the basement membrane (BM), a thin but robust sheet of fiber-structured proteins (e.g. collagens, fibronectins). In healthy lungs, the thickness of the BM measures 5–7 µm, while in asthmatics the size can be found increased up to 5 times [29, ; compare Fig. 1C, 1D]. A multitude of cell types present in the lung can potentially contribute to BM thickening through a distorted deposition of extracellular matrix (ECM) components. Thickening of the asthmatic BM is due to increased deposition of collagen I and III, tenascin, and fibronectin, likely produced by activated myofibroblasts [30, 31]. Several studies demonstrated that the BM is thickened, even in young asthmatic children and this thickening is comparable to that observed in adults [15, 24, 32, 33]. The increased BM thickening has been shown to be correlated with ASM cell mass, indicating that alterations in ECM composition and subsequent BM thickening may be dependent depend on the activity of ASM cells [34]. Furthermore, the ECM is a reservoir for secreted factors, which can be released upon degradation/reconstruction of the ECM within the tissue. VEGF for instance binds to the ECM through heparan sulphate proteoglycans and is released upon matrix degradation processes [35, 36]. When airway remodeling was reported for the first time it was described as a thickening of the BM [1]. Decades after this first description of BM thickening electron-microscopy revealed that the thickened layer is not the BM itself but a zone adjacent to it – the lamina reticularis [37]. Among chronic inflammatory lung diseases, which are also accompanied by ECM alterations (e. g. cystic fibrosis or chronic obstructive pulmonary disease (COPD)) BM thickening – including alterations in dimension and composition – is a unique pathology of asthma [38]. The thickening of the BM has also impact on the efficacy of treatment, as it was correlated with a limited short-term responsiveness to GC treatment [39].
Traveling further down the airway wall, we now enter the submucosa. This layer predominatly exists of connective tissue (CT) and mesenchymal cells, in particular fibroblasts (Fig. 2). Normally, the submucosa is scarsely vascularized and only few micro vessels are observed. In contrast, the submucosa of asthmatic patients is characterized by a marked intensification of vascularity, as well as an increased presence of eosinophils and mast cells (compare Fig. 1A, B). In the asthmatic lung, eosinophils have long been recognized as important immunomodulatory cells involved in airway wall remodeling. Indeed, depletion of eosinophil-derived cytokines reduced deposition of ECM proteins in the subepithelial BM of mild atopic asthmatics [40, 41]. Furthermore, eosinophils produce TGF-β1 which promotes proliferation of fibroblasts, as well as myo-fibroblast maturation and collagen synthesis [42]. Eosinophils and mast cells produce several angiogenic factors, including VEGF, and may thus help to direct angiogenesis in the asthmatic submucosa [43-46].
Travelling still further down the airway wall, we finally touch the most striking feature of the asthmatic airway wall: the distinctive increase of the bulk of ASM cells [47, 48; compare Fig. 1A, B]. Smooth muscle cells, which surround the airways from the trachea down to the alveolar ducts, are the effector cells that determine the diameter of the airways. These are the cells that make the airways to relax or to constrict. Bronchoconstriction is the most severe symptom of an asthma attack and ASM cells are the major effector cells and the proximal cause of the excessive airway narrowing [49]. The constriction is completely or partially reversible, but sometimes only with the help of bronchodilating drugs. Two features of ASM cells are characteristic for asthma patients. First, we observe airway hypersensitivity, which refers to the increased response of ASM cells to low doses of stimuli. Second, the ASM cell of asthmatics demonstrates hyperreactivity, which describes an abnormal strong bronchoconstrictive response. Together, hypersensitivity and hyperreactivity of the ASM cell add up to the well-known asthma-characteristic airway hyperresponsiveness (AHR) [50], AHR can be induced by a plethora of environmental and/or systemic alterations, including allergen-challenge, sudden temperature changes, reduced humidity, exercise, increased inflammatory mediators, viral infections, etc. The bio-molecular cause for AHR remains unclear, however, although it may be due to fundamental changes within the ASM cell [51-53]. It should be noted, however, that the asthmatic ASM cell itself is not contracting stronger. Rather, the increased number of ASM cells (hyperplastia) causes a faster contraction relative to healthy individuals, in addition to an impaired relaxation [54, 55]. The increased ASM mass is already present in young adults (17-23y) [56] and in children without any signs of eosinophilic inflammation [12, 57]. This suggests that the increased number of ASM cells may be the cause rather then the consequence of progressing disease. In addition, ASM cells are highly productive cells, in the sense that they produce a variety of inflammatory and angiogenic mediators, including GM-CSF, IL-1β, IL-2, IL-5, IL-6, IL-8, IL-11, IL10, Eotaxin, bFGF, PDGF-BB and VEGF [58]. In addition, ASM cells of asthmatic patients have a distinct hyper-reactive (“primed”) phenotype, which is characterized by an increased release of pro-inflammatory factors and mediators. For instance, ASM cells of asthmatics release more IL-6 and IL-8 after stimulation with house dust mite (HDM) extracts or after rhinovirus infection [51, 52]. The release of many of the aforementioned cytokines can be induced by several stimuli or in paracrine and autocrine regulated mechanisms. This shows that ASM cells of the airways provide a mechanism how inflammation may be amplified and/or perpetuated without the involvement of immune cells. Furthermore, ASM cells are an important source of ECM components. ASM cells incubated with serum obtained from asthma patients demonstrated an increased production of fibronectin, lamin-γ, and proteoglycans [53, 59, 60]. In addition, ASM cells from asthmatics produce more connective tissue growth factor (CTGF), which may further contribute to BM thickening (as described above) [61, 62]. It is also of note that the number of mast cells in deeper regions of the airway wall is markedly increased and it has been suggested that mast cells are involved in “programming” (or “priming”) the adjacent ASM cells of asthmatics [63-67]. In contrast to the mast cells present in the submucosa, the mast cells located in the ASM bundles are always tryptase and chymase positive (MCTC) and their number has been linked to the severity of asthma [68] and airway hyperresponsiveness [63]. Mast cells are thought to be a major source of angiogenic factors in the lung and the number of tryptase-positive mast cells has been correlated with angiogenesis in human endometrial cancer [69].
ANGIOGENESIS
Angiogenesis refers to the formation of new blood vessels from endothelial cells, a complex process involving multiple factors with either promoting (angiogenic) or counteracting (angiostatic) actions.
In diseases associated with a pathological increase in vasculature, such as the submucosa of asthma patients, the net balance must be shifted towards pro-angiogenic factors. This can be accomplished through a reduction of angiostatic factors, the increase of angiogenic factors or a combination of both. The first report that vascular abnormalities occur in the airways of asthma patients was published in 1960, when Dunhill and coworkers reported the dilatation of blood vessels in lung tissue from asthma patients [70]. Currently, it is well-documented that the sub-epithelial submucosa of the lungs of asthmatic patients displays an increased vascular density – both ex vivo and in vivo [24, 71-73]. In addition, several abnormal modifications of the bronchial vasculature have been described in the asthmatic airways. In asthmatic humans, as well as in animal models of asthma, the blood flow in the lung is increased [74-77]. Morover, the vessels present in the submucosa of asthmatics are more permeable [78]. This not only leads to edema, it is also a prerequisite for tissue inflammation, as the extravasation of inflammatory cells to the tissue requires transmigration through the endothelium.
Increased vascularization has been shown to correlate to airflow limitation [71, 79, 80] and bronchial hyperresponsiveness [80-82], hence contributing significantly to the pathology of asthma. Additional studies provided further evidence that vascular density and asthma severity are linked [46, 83-85]. Furthermore, in the submucosa of asthma patients increased levels of VEGF, bFGF and angiogenin have been observed [86-88]. Simcock and coworkers demonstrated that human ASM cells of asthmatic patients produced higher levels of angiogenin, angiopoietin, VEGF, EGF, IGF-1, IFNγ, TIMP-1 and TIMP-2 in response to stimulation with either IL-13 or TGF-β [86, 87]. Recent studies in our laboratory demonstrated that human ASM cells of asthma patients also released significanly more VEGF, angiogenin, IL-6, MCP-1 and importantly three CXCR2 ligands, namely ENA-78, GRO-α and IL-8 [89].
CXC-chemokines and their receptors are responsible for the progression of many solid tumors, including breast cancer, bronchogenic carcinomas, and malignancies of the gastrointestinal tract, prostate cancer, ovarian cancer, glioblastoma, as well as head and neck cancer [90-95]. CXC-chemokines are also involved in several lung diseases, which involve altered angiogenesis such as idiopathic pulmonary fibrosis, obliterative bronchiolitis, or acute respiratory distress syndrome [96-98]. Comparing the angiogenic potential of ASM cells of asthmatic patients to those of non-asthmatics, we demonstrated that ASM cells of asthmatic patients promote angiogenesis through elevated secretion of the CXC-chemokines ENA-78, GRO-α and IL-8 [99]. Endothelial cell sprouting was significantly increased upon incubation with culture supernatants of ASM cells of asthma patients relative to those of non-asthmatic control subjects and that these effects were solely mediated via the CXCR2 receptor [99; Fig. 3]. Apparently, asthmatic ASM cells exhibit an increased potential to synthesize CXCR2 ligands and may hence help directing the neovascularization in the submucosa of the lungs of asthma patients. Elevated levels of ENA-78, GRO-α and IL-8 have indeed been reported to be present in the BALF/sputum of asthmatic subjects [99, 100]. The increased angiogenic potential of ASM cells of asthma patients may thus explain the increased vascular density in the submucosa observed in the airways of asthma patients. These findings supplemented previous studies showing that ASM cells are a major source of angiogenic factors and that CXCR2 ligands are elevated in the airway lining fluids of asthma patients [86, 87]. Studies in several model organisms (mice, sheep, and primates) showed that HDM allergens increased vascularity of the lungs [101-104]. HDM allergens therefore directly contribute to airway wall remodeling via a non-immune cell mediated mechanism. These data support earlier findings that HDM exerted its effect independently of the immune system via resident tissue-forming cells [105]. Interestingly, the CXC-chemokine ENA-78 is selectively degraded by the proteolytic activity present in HDM extract [106]. Since HDM extract did not have a direct effect on the angiogenic potential of ASM cells, the angiogenic effects might be mediated by immune or endothelial cells, which may release angiogenic mediators upon stimulation with proteases present in HDM excrements [105]. Because ENA-78 acts primarily on neutrophils, altered ENA-78 levels in the lungs may not only affect angiogenic, but also inflammatory responses in the airways of asthma patients. Data on neutrophil transmigration indeed suggested a diminished migration of neutrophils when ENA-78 was incubated with HDM extract [106]. Therefore, the presence of proteases and/or HDM allergens in the airways may alter the bio-availability of ENA-78 and modulate both angiogenic and immune responses. Similarly, a disturbed release off endogenously produced metalloproteinases from activated endothelial cells may affect the turnover of vessel-forming proteins [107], and influence the angiogenic process. Specifically, ADAM33 is thought to play a role in airway remodeling because of its high expression in epithelium, myo/fibroblasts, and airway smooth muscle cells and its role in promoting angiogenesis and stimulating cell proliferation and differentiation [108]. In vivo, the key trigger to induce angiogenesis is hypoxia, i.e. the diminished availability of oxygen in organs and tissues. Hypoxia is observed in many physiological conditions, including embryonic development and differentiation or wound healing. In addition, hypoxia occurs as the consequences of several pathologies including tumor progression, interstitial lung diseases, arteriosclerosis, COPD and asthma [109-111]. Hypoxia of the airways can be caused by a range of mechanisms, including diminished ventilation drive, airway obstruction, intra-alveolar exudates, airway wall inflammation, in addition to fibrosis and basement membrane thickening [112]. That the ASM cells of asthma patients is consuming more oxygen is reflected in the increase of mitochondria [113], a phenomenon that may further add to locally restricted hypoxia. Moreover, an increased ASM mass needs more nutrients and oxygen, hence more vessels. The increased vascularization in the submucosa may contribute to asthma pathology in several ways. First, increased vessel leakage may cause tissue edema and thereby narrowing of the airways. Second, plasma exudation may create and/or aggravate local inflammation and remodeling. Third, an increased blood supply provides the hyperplastic and hypertrophic ASM cells with the required nutritions and oxygen. Reducing the vascularization may therefore counteract both airway wall remodeling and inflammation.
Hypoxia is always associated with the activation of proteins of the hypoxia inducible factor (HIF)-family. The activation of the HIF-pathway allows the cells to adapt to external factors (in this case the low oxygen concentration). The major component of hypoxia signaling is the transcription factor HIF, which is comprised of two subunits (HIF-α and HIF-β). The HIF-1β subunit is constitutively expressed and dimerizes with one out of its three potential binding partners HIF-1α, HIF-2α or HIF-3α. Under normal oxygen conditions (normoxia), the HIF-α proteins have a very short half-life and are, by hydroxylation of two proline-residues in the oxygen-dependent degradation domain, constantly targeted for proteasomal degradation [114]. Ubiquitinylation of hydroxylated HIFα is mediated by the Von Hippel-Lindau protein (VHL) [115]. Under low oxygen concentrations hydroxylation is impaired and results in increased levels of HIF-α. HIF-α is then translocated to the nucleus, dimerizes with the HIF-β subunit and the active transcription factor binds to hypoxia response elements in the promotor regions of downstream target genes [113]. Currently, approximately 100 targets genes of the oxygen-sensing pathway are known and their corresponding proteins are implicated in tissue angiogenesis, erythropoiesis ventilation, glycolysis, and autophagy [116]. A simplified scheme of the HIF-dependent pathway is presented in Fig. (4).
Only recently, a role for hypoxia in the pathogenesis of asthma has been appreciated. For oxygen supply, ASM cells depend on either diffusion though the basement membrane or oxygen delivered by microvessels present in the airway wall. The airway wall of asthmatics is characterized by inflammation and an increased bulk of ASM cells: highly active, oxygen consuming tissue. Furthermore, local hypoxic conditions may occur due to increased thickness and altered composition of the basement membrane, as well as of edema, which may affect the rate of oxygen diffusion [117-119]. The observed increased thickness of the basement membrane of asthma patients may induce a locally restricted hypoxic environment. In addition, increased oxygen consumption, for instance by inflammatory cells and growing BSMC may further add to hypoxic conditions in the asthmatic lung [117]. Together, these conditions may affect the properties and behavior of BSMC, for instance through the induction of HIF. In a rat model of asthma, expression of the transcription factor HIF was indeed associated with airway inflammation, whereas elevated levels of HIF have been detected in endobronchial biopsies and in the bronchial alveolar lavage fluid of asthmatics [110, 120]. Furthermore, increased levels of HIF subunits (HIF-1α and HIF-1β) were found in lung tissues and epithelial cells of asthmatic patients and correlated with VEGF [120]. Recently, we showed that hypoxia significantly reduced the proliferation of BSMC of both asthmatic and non-asthmatic subjects, but with differential responses [121]. Under moderate hypoxic conditions (5% O2), only asthmatic ASM cells significantly reduced proliferation, leaving that of non-asthmatic control cells unaffected. Stringent hypoxic conditions (1% O2) significantly reduced the proliferation of ASM cells from both asthmatics and non-asthmatics. The reduced proliferation under stringent hypoxic conditions (1% O2) was confirmed by lower levels of PCNA- and cyclin E proteins in BSMC of asthmatic and non-asthmatic subjects [121]. This shows that ASM cells of asthmatics are more prone to hypoxic conditions than their non-asthmatic counterparts. The reason for this difference is unclear, but it might reflect diminished proliferation control responses as observed in cultured asthmatic BSMC [51, 53]. In addition, we showed that hypoxia enhanced the release of VEGF, and the angiogenic cytokines IL-6 and IL-8 in ASM cells, with no significant differences between asthmatics and non-asthmatics [121]. It is unlikely, therefore, that the increased vascularization of the submucosa, as it is observed in the airway wall of asthma patients, can be ascribed to the release of these factors under hypoxic conditions. The finding that CXCR2 are important mediators of angiogenesis may provide an additional mechanism how angiogenesis in the asthmatic airway wall can be induced independently of VEGF.
FUTURE PERSPECTIVE
Once thought to be merely an immunological disorder, asthma is increasingly recognized as a disease of the airway wall with the ASM cells as a major player in inflammatory and remodeling responses. Persistent airway wall remodeling is now a well-established pathology of the asthmatic lung and the major player may well be the ASM cell. Relative to their healthy counterparts, ASM cells of asthma patients release more cytokines and chemokines, as well as increased levels growth and angiogeneic factors (Fig. 5). This suggests that the ASM cells of asthma patients are hyper-active (or: “primed”) and may thus induce or aggrevate airway wall remodeling. This “priming” may also be reflected in the lack of efficacy of standard asthma medication in counteracting the airway wall remodeling.
Usually, asthma is treated with beta-mimetics, with glucocorticosteroids (GC), or with a combination of these drugs. Beta-mimetics induce the relaxation of ASM cells, hence reducing bronchoconstriction, whereas GC are very effective at reducing airway inflammation. Despite the benificial anti-inflammatory actions of GC, it remains controversial whether GC are also able to reduce or reverse airway wall remodeling. Recent studies indicate that only high doses of inhaled GC can reduce some components of airway remodelling, such as the increased airway wall vascularity and the basement membrane thickness [122-124]. In contrast, GC do not affect ASM cells proliferation and associated remodeling [125]. Due to lack of specific targets, we currently lack an adequate, non-invasive treatment of the “primed” ASM cell. But this may change soon. Recently, we performed large-scale profiling analyses of signalling pathway activation signatures in ASM cells of asthmatic and non-asthmatic subjects [126]. Of the 270 studied signaling pathways, we observed that a substantial fraction of them was significantly more active in asthmatic ASM cells, so that ASM cells of asthmatic patients can be clearly distinguished from those of non-asthmatics. Importantly, the identified pathways predominantly represented pathways linked to cell proliferation and growth, vascularization, inflammatory response and muscle-tissue specific responses, such as contractility. This study was the first to provide clear-cut evidence for an overall, intrinsic “primed” status of asthmatic ASM cells and may reflect the genetically predisposition of the disease [126].
The realization that the “primed” ASM cells may be pivotal to regulating and controling both inflammation and remodeling of the airways may provide an entry to completely unexplored fields of asthma treatment. Notably, if the airway remodeling – not the inflammation – is the primary etiological event leading to asthma as suggested by studies in asthmatic children [10-17], then asthma prevention might become possible by specifically treating the airway smooth muscle cells, averting them to become hyper-active, hypertrophic and hyperplastic. Indeed, a new, invasive therapy option that specifically addresses the ASM cells has already been used in cases of uncontrolled asthma: thermoplasty. This treatment involves locally restricted heating of the airway wall using radiofrequency energy and significantly reduces the mass of ASM [127]. Thermoplastic reduction of ASM cells reduced the frequency of asthmatic exacerbations [128-133], and in several cases of severe asthma, thermoplastic treatment led to a marked reduction of asthma symptoms and a stable lung function over a long period of time [131]. The efficacy of thermoplasty further emphasizes the role of ASM cells in the pathogenesis of asthma, which makes the search for non-invasive medication and therapeutic strategies that reduce – or even completely abrogate – the remodeling processes worthwhile. For instance, the increased angiogenic potential of ASM cells of asthma patients might be reduced by antagonizing or blocking the CXCR2 receptor or its ligands. Studies with CXCR2-blockers and ligand-neutralizing agents in the context of different diseases, such as rheumatoid arthritis and COPD, are ongoing [129, 134]. In vitro, we showed that this strategy abrogated the increased sprout outgrowth from EC spheroids induced by super natants of asthmatic ASM cells [89]. CXCR2 also mediates the recruitment of endothelial progenitor cells during allergic airways remodeling [135]. Thus, the CXCR2-mediated neovascularization may be targeted using extant pharmaceutical compounds and may represent a novel strategy to reduce airway remodeling in asthma. A more profound discussion on the outcomes of recent clinical trials with anti-angiogenic therapies and their potentially asthma-resolving properties was recently provided by Harkness and coworkers [136].
In conclusion, a fundamental demand exists to develop new treatment strategies to tackle the airway wall remodeling observed in the lungs of asthma patients. The discussed studies demonstrate a pivotal role for a “primed” ASM cell in the airway wall remodeling process. Not only do they have an increased proliferative capacity, they also produce increased levels of growth factors, chemokines and cytokines, as wel as angiogenic factors. Targeting the “primed” ASM cell to reduce its activity may act as a double edged sword. First, reduced numbers of ASM cells will lead to diminished airway contraction, hence decreasing the bronchi to constrict. Second, reduced numbers of “primed” ASM cells will reduce the inflammatory and angiogenic mediators in the airway wall and concomitantly reduced vascularization. The latter may have profound beneficial effects. Blood vessels are the primary portal for immune cells to infiltrate tissues and the increased number of blood vessels in the airway wall of asthma patients might lead to exaggerated inflammation [137]. There is evidence that badly controlled asthma results in an increase in inflammatory cells, which are entering the airway tissue via ancillary blood vessels [138]. Here too, both ASM cells and the endothelium might be promising novel targets to reduce airway wall remodeling and asthma symptoms. The development of (inhaled) anti-angiogenic agents might be an interesting new strategy to reduce smooth muscle mass. In the fight against cancer, the importance of angiogenesis has been recognized and several anti-angiogenic drugs have already been positively tested in clinical trials. Developers of future asthma therapies may as well look into that direction.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
LFK and PB were supported by a research grant of the Swiss National Foundation (#320030_124905/1).

