All published articles of this journal are available on ScienceDirect.

Pathophysiological Approaches of Acute Respiratory Distress syndrome: Novel Bases for Study of Lung Injury
Abstract
Experimental approaches have been implemented to research the lung damage related-mechanism. These models show in animals pathophysiological events for acute respiratory distress syndrome (ARDS), such as neutrophil activation, reactive oxygen species burst, pulmonary vascular hypertension, exudative edema, and other events associated with organ dysfunction. Moreover, these approaches have not reproduced the clinical features of lung damage. Lung inflammation is a relevant event in the develop of ARDS as component of the host immune response to various stimuli, such as cytokines, antigens and endotoxins. In patients surviving at the local inflammatory states, transition from injury to resolution is an active mechanism regulated by the immuno-inflammatory signaling pathways. Indeed, inflammatory process is regulated by the dynamics of cell populations that migrate to the lung, such as neutrophils and on the other hand, the role of the modulation of transcription factors and reactive oxygen species (ROS) sources, such as nuclear factor kappaB and NADPH oxidase. These experimental animal models reproduce key components of the injury and resolution phases of human ALI/ARDS and provide a methodology to explore mechanisms and potential new therapies.
1. INTRODUCTION
1.1. Definition of Acute Respiratory Distress Syndrome
Acute respiratory distress syndrome (ARDS) is defined as an acute-onset, progressive, hypoxic condition with radiographic bilateral lung infiltration, which develops after several diseases or injuries, and is not derived from hydrostatic pulmonary edema. One specific pathological finding of ARDS is diffuse alveolar damage. In 2012, in an effort to increase diagnostic specificity, a revised definition of ARDS was published in JAMA [1]. The acute respiratory distress syndrome (ARDS) was defined in 1994 by the American-European Consensus Conference (AECC) [2]; since then, issues regarding the reliability and validity of this definition have emerged. Using a consensus process, a panel of experts convened in 2011 (an initiative of the European Society of Intensive Care Medicine endorsed by the American Thoracic Society and the Society of Critical Care Medicine) developed the Berlin Definition, focusing on feasibility, reliability, validity, and objective evaluation of its performance. A draft definition proposed 3 mutually exclusive categories of ARDS based on degree of hypoxemia: mild (200 mm Hg < PaO2/FIO2 ≤ 300 mm Hg), moderate (100 mm Hg < PaO2/FIO2 ≤ 200 mm Hg), and severe (PaO2/FIO2 ≤ 100 mm Hg) and 4 ancillary variables for severe ARDS: radiographic severity, respiratory system compliance (≤40 mL/cm H2O), positive end-expiratory pressure (≥10 cm H2O), and corrected expired volume per minute (≥10 L/min). The draft Berlin Definition was empirically evaluated using patient-level meta-analysis of 4188 patients with ARDS from 4 multicenter clinical data sets and 269 patients with ARDS from 3 single-center data sets containing physiologic information. The 4 ancillary variables did not contribute to the predictive validity of severe ARDS for mortality, and were removed from the definition. This updated and revised Berlin Definition for ARDS addresses a number of the limitations of the AECC definition [1]. The approach of combining consensus discussions with empirical evaluation may serve as a model to create more accurate, evidence-based, critical illness syndrome definitions and to better inform clinical care, research, and health services planning.
However, no new parameters or biomarkers were adopted by the revised definition. Discriminating between ARDS and other similar diseases is critically important; however, only a few biomarkers are currently available for diagnostic purposes. Furthermore, predicting the severity, response to therapy, or outcome of the illness is also important for developing treatment strategies for each patient. However, the PaO2/FIO2 ratio is currently the sole clinical parameter used for this purpose. In parallel with progress in understanding the pathophysiology of ARDS, various humoral factors induced by inflammation, and molecules derived from activated cells or injured tissues have been shown as potential biomarkers that may be applied in clinical practice.
This review is based on the pathophysiologic of ARDS and how the discovery of new homeostatic control and progression mechanisms have changed the development of experimental models.
2. PATHOPHYSIOLOGY OF ARDS
ARDS was recognized as the most severe form of acute lung injury (ALI), a form of diffuse alveolar injury. The ARDS is defined as an acute condition characterized by bilateral pulmonary infiltrates and severe hypoxemia in the absence of evidence for cardiogenic pulmonary edema. [3]. The mechanisms that initiate the lung injury have not been totally understood, despite almost 35 years of full investigation. Clinical trials have provided relevant information about the onset and evolution of the physiological and inflammatory variables in the lungs. This information has led to hypotheses about pathophysiology of injury, but for the most part, this paradigm have been difficult to test in humans due to the difficult to control of the clinical variables in critically ill patients.
During the pathophysiological sequelae of the human ARDS, the increase of capillary permeability due to the decrease of blood oxygen saturation (shock syndrome) leads to a vascular leakage (exudative phase) [4]. This leakage becomes evident in an increasing perivascular and intraalve-olar edema with transudation of soluble, noncellular blood factors like fibrin into the lung alveoli. Therefore, previous histopathological assessment of the human situation showed as one dominating feature of ARDS, a perivascular edema, which primarily was found in the central parts of the lung of a patient who died 5 min after an accident. An intrapulmo-nary, intravascular accumulation of polymorphonuclear granulocytes was detectable in humans after 90 min and the edema at this time point was generalized in the whole lung [5]. The formation of perivascular and intraalveolar edema was accompanied by an increase in lung weight, which is significantly correlated with the survival time of humans in ARDS [6].
Currently, there is a major advance in the knowledge of the pro-inflammatory molecular mechanisms of these events, including the contribution of environmental and genetic factors. In detail, ALI/ARDS is characterized by an initial insult that triggers cell-mediated mechanisms releasing a cascade of a variety of mediators, which are illustrated in Fig. (1) and discussed in detail below.
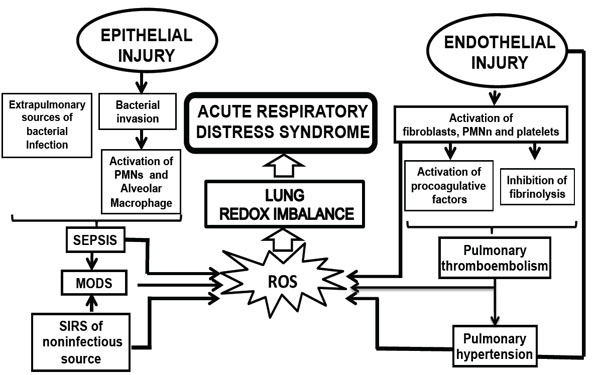
Pathophysiology events related with the develop of ARDS. MODS, multiorganic; PMNn, polymorphonuclear neutrophils; ROS, rective oxygen species.
Endothelial damage. In the acute or exudative phase of ALI/ARDS, protein-rich edema fluid enters the air space as a result of the increased permeability of the alveolar-capillary barrier [7]. This increased permeability leads to the formation of pulmonary edema. In addition, serum proteins precipitate with cellular debris in the alveolar spaces, forming the characteristic hyaline membranes.
Epithelial cell damage. The loss of epithelial integrity induces alveolar flooding and impedes the removal of fluids from the alveolar space. Injury of cuboidal type II cells alters ion transport, fluid removal capacity and surfactant production, and triggers the release of various mediators, including proinflammatory cytokines, leading to the activation of neutrophils and stimulating chemotaxis [8]. In addition, damage at this level can induce and facilitate bacterial invasion leading to sepsis and septic shock.
Activation of cytokines, neutrophils and macrophages. Triggering of cytokines initiates and amplifies the inflammatory response. The inflammatory process may originate from a direct insult, such as pneumo-nia, aspiration, or inhalation of substances, or second-arily as part of a systemic inflammatory response, such as in sepsis or trauma. Excessive recruitment of leukocytes is relevent to the pathophysiology of ALI, and the magnitude and the time of presentation of the inflammatory events associated with the clinical prognosis in patients with ALI. Leukocyte recruitment is a coordinated mechanism that depends on the chemotactic process and chemokine receptors [9]. Understanding the cellular events that are associated with the leukocyte recruitment may lead to the design of effective therapeutic approaches.
The roles of proinflammatory cytokines such as tumor necrosis factor and interleukins-6 and IL-8 have been extensively studied, and these cytokines have been associ-ated with the development of ALI/ARDS in several models, with complex relationships of induction and regulation elucidated with other substances, such as endotoxins and the proinflammatory transcriptional factors, such as nuclear factor kappaB (NF-(κB) [10]. In addition, cytokine product-ion has been shown to vary according to the origin of ARDS (e.g., in trauma versus sepsis).
2.1. Role of Polymorphonuclear Neutrophils in ALI/ ARDS
The presence and activation of neutrophils in ALI/ARDS is well established, given the presence of polymorphonuclear neutrophils (PMNs) and their products, such as lysosomal enzymes in bronchoalveolar lavage from patients with ARDS, although lung injury can develop in the absence of neutrophils, such as in neutropenic patients.
In the early stage of ALI, PMNs attach and are recruited to the pulmonary capillaries, and then migrate into alveolar spaces where PMNs are activated and release cytotoxic substances (oxygen free radicals, lipid mediators, and proteases) resulting in injury of alveolar epithelial cells and capillary endothelial cells. The recruitment of PMNs into areas of inflammation is a complex process involving PMN tethering, capturing, rolling, activation, and firm adhesion. Inflammatory mediators are involved in each step. The recruitment of PMNs in pulmonary microvessels is one of the earliest events in patients with ALI/ARDS [11], which first appears as a change in cell biomechanics and kinetics. Because the diameter of pulmonary capillaries is less than the diameter of PMNs, these cells can only emigrate out of capillaries by deformation and elongation. However, because the corresponding receptors on the surface of PMNs are stimulated by inflammatory mediators and intracellular signaling molecules (Ca2+) are activated, the cytoskeleton of PMNs redistribute, which leads to PMNs stiffening and a decrease in deformation, resulting in a large number of PMNs gathering in the capillary and a reduction of PMNs in the peripheral blood of patients with ALIs [12, 13]. In humans, PMNs pool in the bone marrow, circulation, and tissues. When inflammatory mediators enter the circulation, the bone marrow is stimulated to release PMNs [12]. It has been demonstrated that PMNs can be released from the bone marrow 7-10 min after complement fragments are injected into the body [14, 15], indicating that PMN recruitment in pulmonary microvessels is due to a change in mature PMN biomechanics and many immature PMNs migrate into the circulation, as deformation of immature PMNs is flawed. However, continuous recruitment of PMNs depends on PMN adhesion to endothelial cells.
Cell adhesion molecules mediate the adhesion between PMNs and endothelial cells activated by the endothelium, in which steps include loose adherence and compact adhesion. Cell adhesion molecules are glycoproteins located on the cell surface and are involved in binding with other cells or the extracellular matrix. Cell adhesion molecules include the cadherins, selectins, immunoglobulins, and integrins [16].
Following endotelial adhesion, PMNs are co-activated by pro-inflammatory agonists and the adhesion surface. The adhesion surface bears the integrin ligand, ICAM-l. An integrin is crucial in the activation of PMNs, which must be stimulated and bind to ligands. Integrin is stimulated by the following two signal transduction methods and another alternative mechanism: outside-to-inside; and inside-to-outside [17]. After binding to ligands, activated integrins stimulate PMNs through a series of complex biological signaling processes. There are three signal transduction pathways. First, the cytoskeleton linker protein phospho-rylation makes a transmembrane integrin link with the actin cytoskeleton and binds to many signal molecules. Src tyrosine kinase is one such signal molecules and is signifi-cant in generation of oxidants during the respiratory burst; Second, the Ras-Raf-MAPK pathway participates in cyto-kine expression and secretion, in which it is crucial that Src tyrosine kinase stimulates transcription factor NF-(κB. NF-κB regulates the expression of immediate early genes involved in the stress response, immune response, inflam-mation, and apoptosis, and is also an upstream transcription factor of ICAM-1 [18]. Activated NF-κB can regulate the expression of ICAM-1, IL-1, and TNF-α [19, 20], which are released from PMNs, adhesión molecules, and endothelial cells after attack by toxins or bacteria in lung tissues. As a result, the microcirculatory disturbances and pulmonary damage are aggravated. Third, PI3K-Akt signaling involves PMN activation and migration through activating NF-κB (Fig. 1).
In summary, PMNs are stimulated by integrins and become high affinity. When integrins bind to ligands and cluster, PMN function is activated [21]. In addition, other molecular pathways also required in the stimulation of PMNs, such as included p38-MAPK phosphorylation. The MAPK family is fundamental in mediating numerous changes in cell function such as cytokine expression, proliferation, and apoptosis [22]. The p38 MAPK, JNK, and ERK play a central role in these cell responses. Upon activation by upstream regulators, the MAP kinases translocate to the cell nucleus where they transform the action of nuclear transcription factors and kinases, which in turn cause changes in cell function such as release of cytokines [23]. This molecular pathway is a therapeutic target that is currently being explored in the inflammatory damage associated with the progression of ALI [24].
2.2. Oxidative Stress as a Signaling Pathway in ALI/ ARDS
ALI/ARDS is characterized by an initial insult that triggers cell-mediated mechanisms releasing a cascade of a variety of mediators, resulting in a redox imbalance among other outcomes [25]. This imbalance is manifested as oxidative stress, and it plays an important role in the development, modulation and perpetuation of the local and systemic inflammatory responses. Although some events during the development of inflammation are common to several organs, the pulmonary parenchyma exhibits special performance with unique features that require a different sort of analysis for the pathophysiology of ARDS/ALI.
The alveolar compartment is permanently exposed to reactive oxygen (ROS) and nitrogen species (RNS) derived from inhaled air pollutants. The antioxidant defense system of the epithelial lining fluid consists of several low molecular weight compounds, such as reduced glutathione (GSH), ascorbic acid (vitamin C) and uric acid; lipophilic antioxidants, such as α-tocopherol (vitamin E), retinol (vitamin A) and plasmalogens (1-alkenyl-phospholipids); and antioxidant enzymes, such as superoxide dismutase, catalase and glutathione peroxidase. Under normal conditions in the lung, complex and coordinated interactions occur among all these antioxidant compounds to provide protection of the distal lung structures from the damaging effects of oxidative attack [26].
As we noted, regardless of the underlying illness, the clinical and pathologic manifestations of ALI/ARDS have similar features, indicating the existence of common final pathways. Therefore, a severe injury caused by an initiating event can lead to dysfunction and a compromise of the barrier properties of the pulmonary endothelium and epithelium as a consequence of an unregulated acute inflammatory response. The earliest events are characterized by the activation of the pulmonary endothelium and macrophages (alveolar and interstitial), up-regulation of adhesion molecules, and production of cytokines and chemo-kines that induce a massive sequestration of neutrophils within the pulmonary microvasculature [27]. These cells transmigrate across the endothelium and epithelium into the alveolar space and release a variety of cytotoxic and pro-inflammatory compounds, including proteolytic enzymes, cationic proteins, lipid mediators, additional inflammatory cytokines and the compounds of interest here: reactive oxygen species (ROS) and nitrogen species (RNS).
The pathogenetic role of reactive species is highlighted by elevated levels of plasma and lung oxidants in patients with ALI/ARDS. In addition, these levels of oxidants correlate with severity of the disease. In animal models of ALI, neutrophil-derived ROS and RNS caused lung injury as shown by histological examination and permeability measurements [28]. Some studies revealed that ROS can disrupt intercellular tight junctions of the endothelium by phosphorylation of focal adhesion kinase [29]. In vitro, ROS induced cell apoptosis and necrosis of alveolar type II cells during oxygen exposure [30]. In vivo, NOX-1−/− mice, but not NOX-2−/− mice, are protected from hyperoxia induced ALI; these results reveal that NADPH oxidase 1 plays a crucial role in hyperoxia-induced ALI [31]. A previous study with NADPH oxidase−/− mice revealed no protection from CVF-induced ALI, indicating that oxygen radical production and lung injury may occur through alternative pathways in mice with genetic deletion of NADPH oxidase [32]. Furthermore inhibition of NADPH or nitric oxide synthase (NOS) has decreased sepsis-induced ALI [33] and respectively LPS induced ALI [34]. Indeed, molecular mechanism included the activation of redox-sensitive transcription factors (NF-kappaB, AP-1), resulting in a large output of proinflammatory cytokines and chemokines, which further aggravate inflammation and oxidative stress. During the process, transcription factor nuclear factor-erythroid 2-p45-related factor 2 (Nrf2) and heme oxygenase (HO) appear to play the counterbalancing roles to limit the propagation of oxidative stress and inflammatory responses in lung [35].
2.3. The Role of Arginase in ALI/ARDS
The function of epithelial and endothelial barriers in the lower zones of the lungs has to be tightly regulated, with intention to prevent leakage and to assure efficient gas exchange between the alveoli and capillaries. Both G− and G+ bacterial toxins, such as lipopolysaccharide and pneumo-lysin, respectively, can be released in high levels within the pulmonary tissue upon antibiotic therapy in patients with ARDS [36]. These toxins alter endothelial barrier function, either directly, or indirectly, by induction of pro-inflammatory mediators and neutrophil activation. Endothelial nitric oxide synthase (eNOS) was proposed to be a relevant part of basal barrier function, since eNOS knock-out mice display an alter expression of inter-endothelial junction proteins and as such an increased vascular permeability, as compared to control mice. The enzyme arginase, the activity of which can be regulated by the redox status of the cell, exists in two isoforms - arginase 1 (cytosolic) and arginase 2 (mitochondrial) - both of which can be expressed in lung structural endothelial cells. Upon activation, arginase competes with eNOS for the substrate l-arginine, as such impairing eNOS-dependent NO generation and induce ROS generation by the eNOS in prooxidant environment [37].
In rat alveolar macrophages, bacterial LPS, released upon antibiotic therapy, was shown to increase both arginase 1 and 2 expression, an effect which could be counteract by glucocorticoids [38]. It is important to highlighted that arginase in alveolar macrophages not only contributes to impairment of NO generation, but it can also determine airway remodeling, as detected in asthma, cystic fibrosis, and chronic obstructive pulmonary disease, through the increased production of l-proline, a precursor of collagen, and the polyamines putrescine, spermidine, and spermine from l-ornitine-derived [39]. The role of arginase in alveolar macrophages in LPS-induced acute lung injury remains controversial, since a recent study showed that mesen-chymal stem cell-conditioned medium mediates the reso-lution of LPS-induced acute lung injury, ameliorates lung inflammation and promoting a wound healing/anti-inflam-matory M2 macrophage cell, includes and arginase 1 activity increases, in an insulin growth factor 1 (IGF-1)-dependent form [40].
3. EXPERIMENTAL APPROACHES IN ALI/ARDS
Experimental approaches have been used to investigate mechanisms of lung damage. Most are support on reproducing in animals known risk factors for ARDS, such as sepsis, lipid embolism secondary to bone fracture, ischemia-reperfusion injury of pulmonary vascular zones, and other risk factors. However, none of these approaches fully reproduces the characteristics of human lung injury.
3.1. Special Issues
3.1.1. Animal Size
Animal size is an important consideration when selecting animal models of ALI. This is especially true when physiological parameters such as arterial oxygen tension and mean arterial pressure are monitored. Until recently, body size has prohibited the measurement of physiological parameters in mice. With the development of small implantable radiotelemetry transmitters, the measurement of systemic blood pressure, heart rate, and temperature is now possible in unrestrained mice, although it remains difficult and expensive [41]. Also, Size is an relevant consideration for the collection of blood. It is easier to obtain sufficient quantities of blood or to obtain multiple blood samples in larger species such as the rabbit or primates.
3.1.2. Species-Specific Reagents
Researchers are often faced with a lack of species-specific reagents to measure inflammatory cytokines, receptors, or other mediators, which are involved in the mechanism of ALI/ARDS. To elucidate this problem, crossspecies reagents have been utilized. In various studies in which comparisons were made between species-specific and crossspecies ELISAs, species-specific ELISAs showed more sensitive and specific data [42].
3.2. Models That Target Primarily the Endothelium
3.2.1. Oleic Acid Administration
Oleic acid is toxic to endothelial cells in vitro [43]. Within 1 min of intravenous oleic acid administration (0.04 mL/kg in dogs), electron microscopy reveals severe vacuolation of endotelial cells, and passage of Evans blue, into the subendothelial space [44]. The actual mechanism whereby oleic acid induces cell death is due to the down-regulation of anti-apoptotic marker Bcl-2 and up-regulation of pre-apoptotic marker Bad, among others [45]. Indeed, there is neutrophilic activation, but the injury is not only related with neutrophil activity, suggesting that a direct effect of oleic acid on the endothelium is the pivotal event [46]. Electron microscopic examination reveals necrosis and detachment of alveolar epithelial cells, necrosis of endothelial cells, and exposure of Hystological examination shows necrosis and breakup of alveolar epithelial cells, necrosis of endothelial cells, and exposure of the basal membrane on both the epithelial and the endothelial border [47]. The gas exchange impairments derived from severe ventilation/perfusion (V/Q) mismatching, increased intrapulmonary shunt, and aerea dead space [48]. The static lung compliance and functional residual capacity fall fastly, and mean airway pressure rises. the oleic acid model produces the fundamental characteristics of ALI, early and quikcly reversible patchy inflammatory lung injury with permeability changes and alteration in gas exchange and lung mechanics. A relevant advantage of this experimental approach is its reproducibility.
3.2.2. Endotoxin Administration
In this model of ALI/ARDS, the intravenous adminis-tration of LPS induces the initial injury in the capillary endothelium. The cellular damage induced by LPS appears to be associated with apoptosis, in contrast to the oleic acid model, in which the injury is due to a direct toxic effect which induces necrosis. Endothelial cells show apoptosis in a short-term following administration of LPS and precedes other tissue injury [49]. Changes in the lungs become observable within 2-4 h and include hypoxemia with an increase in the alveolar-arterial oxygen difference (A-a O2 difference). In the lungs, the administration of LPS, either by intravenous In the lungs, the intravenous administration of LPS, either by or intra-alveolar, derived in changes in neutrophil deformability and the neutrophil sequestration in the pulmonary capillaries [50]. However, only small numbers of neutrophils migrate into the air spaces. Polymorphonucler entrapment occurs before epithelial permeability changes or disruption of the alveolar membrane. The pharmacological advantage of the use of LPS in ALI/ARDS is that is easy to administered, and the data tend to be reproducible within experiments. LPS is a potent inductor of the innate immune responses via TLR4 pathways and has minor direct toxicity to cells in vitro [51]. Thus, the use of LPS provides mechanisms about the effects of host inflammatory responses, which occur in bacterial infections.
3.3. Models That Target Alveolar Endothelium
3.3.1. Hyperoxia Exposure
In different mammalian species, exposure to 100% O2 could derive in respiratory distress and lethal multiorgan failure [52]. However, the similar findings have not been reproduced in humans with normal lungs, who develop only mild increases in alveolar capillary permeability after 24 h of exposure to 100% oxygen, and no clinical or hystological result of lung injury when the time of exposure is up to several days [53]. Oxygen-based approaches derived in the lung elevated ROS levels and induce lung cell injury and inflammation. Injured cells are replaced through increased proliferation and differentiation of epithelial cells and fibroblasts [54].
Hyperoxia-induced lung injury can be determine as a bimodal process resulting from direct oxygen toxicity [55] and from the accumulation of inflammatory cells and mediators within the lungs. ROS can damage the lung cells directly, mainly through lipid peroxidation leading to cellular membrane disruption, followed by cellular necrosis. It is known that hyperoxia results in leakage of ROS from the mitochondrial complexes, from the process of hypoxia/reoxygenation, whereby the conversion of xanthine dehydrogenase to oxidase, and from activated neutrophils recruited to the lungs through NAPDH superoxide production. In addition, ROS has been shown to be involved in the recruitment of inflammatory cells by inducing prolonged expression of specific neutrophil that interact with endothelial cell surfaces in a long-term form, for example Endothelial Leukocyte Adhesion Molecule-1 [56] and Granule Membrane Protein-140 [57, 58].
3.3.2. Surfactant Depletion
Diminution of surfactant may be determine lung injury by two mechanisms: first, due to alveolar collapse and increasing the likelihood of mechanical injury to the alveolar walls during repeated cycles of opening/closure related to invasive mechanical ventilation, and second, through alteration of alveolar host defenses. The administration of bovine surfactant and/or synthetic surfactant-related components attenuates the epitelial damage induced by saline lavage [59, 60]. Saline lavage (0.9% NaCl) leads to almost immediate hypoxemia and widening of the A-a O2 difference. These findings are fastly corrected by recruitment maneuvers such as sustained inflation, suggesting that gas exchange alterations reflect the collapsed alveoli with otherwise intact alveolar walls [61]. The saline lavage by itself has few consequence on permeability due to inflammatory changes [62].
Surfactant dimunution is an relevant characteristic of ALI/ARDS in humans, but unlike newborns, surfactant depletion in adults is commonly a consequence rather than a primary effect associated with of lung injury [63]. In ARDS, surfactant abnormalities occur due to the injury of the alveolar epithelium and exudation of protein-rich edema fluid into the alveolar spaces. Saline lavage of the lungs results in surfactant depletion in the absence of the increase of damage in alveolar epithelial.
3.4. Models Targeting the Epithelium and Endothelium
3.4.1. Ischemia/Reperfusion Models
A classic form of injury secondary to lung I/R is the “reimplantation response,” a type of tissue injury following lung transplantation. The reimplantation response is characterized by non-cardiogenic pulmonary edema, inflammatory infiltrates, and hypoxia events which are unrelated to rejection [64, 65]. The pathophysiology of IR injury included two phases, which are expressed during the development of the experimental models: ischemic and reperfusion. In experimental approaches, ischemia can be developed in different form to obstruct pulmonary circulation [66]. In every IR model, two major variables need to be considered during the ischemia phase: (1) the extent of the ischemic bed (pulmonary, bronchial circulation, venous return, and ventilation) as well as (2) the duration of ischemia. In general, lungs have three sources of oxygenation: pulmonary arteries, bronchial arteries, and alveolar ventilation. The extent to which each of these sources shares in providing oxygen for the lung tissue, especially during the IR, has not yet been established. Additionally, two scenarios exist for inducing ischemia in the lungs: “ventilated ischemia” occurs when the blood circulation in the lungs is blocked and ventilation the area of ischemic contribution (ie. pulmonary, bronchial circulation); (2) the time of duration of ischemia. Oxygen supply to lung is provided from different sources: pulmonary arteries, bronchial arteries, and alveolar ventilation. The extent to which each of sources providing oxygen for the lung structure, especially during the ischemia and reperfusion, has not been fully dilucidated. Indeed, two events are described during ischemia in the lungs: “ventilated ischemia” occurs when the blood circulation in the lungs is interrumped and ventilation continues, and “ischemic anoxia” occurs when the blood flow and ventilatory process are blocked. The anoxic ischemia can be developed in the inflated or deflated lung [67]. Indeed, Luh et al. [68] suggested that the lung injuries would be significant if the pulmonary perfusion and ventilation were blocked for >60 min. Among others, the reperfusion phase in the lung is determinant to pathophysiological events, such as gas exchange imbalances.
The effect of a variability of ischemia and reperfusion time on lung damage is still under discussion. In a sheep, an ischemic period after 15–30 min is associated with hyperpnea with intercostal retraction development in this model. Lung functional impairment was shown in other approaches with ischemic time of 30 min without reperfusion, or more prolonged ischemic times [67].
Among others, lungs need the reperfusion phase to indicate the physiological dysfunction, such as fluid and gas exchange imbalances [69]. As observed in the microstructural level, the lung tissue can be damaged more by reperfusion compared with ischemia [70]. Regardless of the structural impairment in the reperfusion phase, certain physiological variables, such as pulmonary arterial pressure and wet-todry ratio, were modified at the early reperfusion time. Reperfusion after ischemia is associated with the developed of leukopenia, fever, and severe ventilation or perfusion mismatch in animal models [71]. Moreover, it was demonstrated that the ROS levels and lipid peroxidation are increase only in the reperfusion phase [72]. At the molecular levels, reperfusion damage is induced by ROS burst, derived from ROS sources induced during ischemic period, such as NADPH oxidase and uncopling nitric oxide synthase [73]. Indeed, IR damage is combined with severe inflammatory response, including neutrophils infiltration in the lung tissue as well as microvascular injuries and edema. These injury pathways resulted in ARDS after some quirurgic maunevers in esophagectomy and in the others similar techniques [74].
3.5. Models of ALI/ARDS secondary to peritonitis
3.5.1. Cecal Ligation and Puncture
Animal models of sepsis have been developed from to intraabdominal infections [75], in which ALI/ARDS would induced by peritonitis. Experimental peritonitis can be produced surgically by ligating and perforating the cecum (cecal ligation and puncture; CLP), or, alternatively, the bacteria can be incorporated into a carrier vehicle such as a fibrin clot or a sponge placed in the peritoneum [76]. The severity of the injury depends on the number of holes in the cecum, and on the size of the needle used to make the holes [77]. In contrast to models using LPS and live bacteria, in which the effects are almost immediate, the effects of CLP develop over some days, and the onset is less abrupt. The main features of CLP-associated lung injury are hypoxemia, neutrophilic inflammation, and interstitial and alveolar edema [78]. Blood cultures are usually positive for multiple organisms, the most common being enteric gram-negative rods such as serratia, enterobacter, and bacteroides species [79]. It is probably the single best animal model of sepsis and organ injury, but the main disadvantage is the requirement for major surgery [80]. In addition, the actual bacterial inoculum in CLP is unknown, and the model could be affected by differences in colonic flora among animals, resulting in significant variability.
The animals models described in ALI/SRS, mechanisms and technical issues are shown in Table 1.
The animal models described from ALI/SRS, mechanisms and procedure issues
| Model | Mechanisms | Procedure issues [REF] |
|---|---|---|
| Oleic Acid | Apoptosis endotelial cells V/Q alterations |
Requires i.v. Inyection [46] |
| Endotoxin (LPS) | Neutrophilic infiltration (A-a) O2 |
Very reproducible [81] |
| Hyperoxia | ROS concentration Shunt |
Requires monitorization gas concentration [82] |
| Surfactant depletion | Alveolar collapse ¯ Lung compliance Hypoxemia |
Animals must be ventilated [83] |
| Ischemia/reperfusion | ROS sources induction Neutrophilic infiltration Edema V/Q alterations |
Complex animal surgery [64, 65] |
| Cecal Ligation & puncture | Permeability interstitial and alveolar edema V/Q alterations |
Biological variability of Surgery [84] |
* LPS, lipopolysacaride; V/Q: ventilation/perfusión; (A-a), alveolar-arterial gradient; ROS, reactive oxygen species.
CONCLUSION
The pathophysiological basis of ALI/ARDS involved an activation and modulation of various inflammatory and inmune events. In this view, the role of host in this evolution are not fully understood. With regards to this review, relevant data has been showed from each of these experimental approaches. However, none of these models adequately reproduces the full characteristics of human ARDS, namely an acute neutrophilic alveolitis with hyaline membrane formation and intravascular microthrombi, and therefore, the choice of a singular model should be made after considering the specific characteristics of each model. Some limitations are common to all approaches of ARDS. Most models are based on one, or at most two methods related with type of injury. However, human ARDS is seldom caused by any single event. More often, ARDS in humans is associated with complex interactions between primary clinical factors such as sepsis and oropharyngeal aspiration; comorbidities, such as diabetes, lung and renal disease; and additional factors inherent to the host, such as inherited genetic factors. Another shortcoming of animal models is that interspecies variability in innate immunity and other responses may affect the importance of animal results for humans extrapolation. Furthermore, the study of molecular markers (inmune or inflammatory molecules), and the clinical value in early detection of severe disease remains to be tested in patients with established severe ALI/ARDS. The specificity of these markers from ALI/ARDS to progression of multiorganic failure, as well as statistical power of their performance will require follow up studies in larger and more diverse clinical cohorts.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Author’s Role: All authors helped to write the manuscript and have seen and approved the final version.

