All published articles of this journal are available on ScienceDirect.

Transfusion-Related Acute Lung Injured (TRALI): Current Concepts
Abstract
Transfusion-related acute lung injury (TRALI) is a life-threatening intervention that develops within 6 hours of transfusion of one or more units of blood, and is an important cause of morbidity and mortality resulting from transfusion. It is necessary to dismiss other causes of acute lung injury (ALI), like sepsis, acute cardiogenic edema, acute respiratory distress syndrome (ARDS) or bacterial infection. There are two mechanisms that lead to the development of this syndrome: immune-mediated and no immune- mediated TRALI. A common theme among the experimental TRALI models is the central importance of neutrophils in mediating the early immune response, and lung vascular injury. Central clinical symptoms are dyspnea, tachypnea, tachycardia, cyanosis and pulmonary secretions, altogether with other hemodynamic alterations, such as hypotension and fever. Complementary to these clinical findings, long-term validated animal models for TRALI should allow the determination of the cellular targets for TRALI-inducing alloantibodies as well as delineation of the underlying pathogenic molecular mechanisms, and key molecular mediators of the pathology. Diagnostic criteria have been established and preventive measures have been implemented. These actions have contributed to the reduction in the overallnumber of fatalities. However, TRALI still remains a clinical problem. Any complication suspected of TRALI should immediately be reported.
1. INTRODUCTION
Transfusion Related Acute Lung Injury (TRALI) is defined as new ALI which develops within 6 hours of transfusion of one or more units, not attributable to another ALI risk factor. Amongst the numerous possible complications, TRALI has emerged as the most important cause of morbidity and mortality resulting from transfusion of blood or derived of the blood components. In fact, TRALI is currently being regarded as the first cause of severe morbidity and mortality related to blood transfusion therapy [1, 2].
The TRALI concept was described in 1985 by Popovsky, who published a trial on surgical patients, who developed hypotension, hypoxemia and respiratory failure, in the absence of hemodynamic overload from 1-6 hours after blood transfusion. A 72 % of these patients required mechanical ventilation. There was a resolution of the condition to 96h of onset of symptoms in 81% of cases. Moreover, the presence of leukocyte antibodies and anti-HLA type I was evidenced in a large percentage of donors [3].
Two decades ago, TRALI was considered a rare complication of transfusion medicine. Nowadays, TRALI has emerged as the leading cause of transfusion-related mortality, presumably as a consequence of reaching international agreement on defining TRALI with subsequent increase recognition and reporting of TRALI cases [4]. The consensus definition in 2004 allowed a better estimate of TRALI, and possible TRALI for populations where other risk factors for acute lung injury are often present, mostly in critically ill patients. Of note, the incidence of TRALI is 50-100 times higher in the critically ill than the general hospital population [2, 5]. In 2004, the consensus conference organized by the Canadian Blood Service and Hema-Quebec (Canada) established the criteria of the Canadian definition, which although very similar to those of the US consensus, introduced the difference between TRALI and possible TRALI, depending on whether or not a temporary relationship to other alternative risk factor for ALI production [6].
Both of them have limitations:
- Only identify new and severe hypoxemia. The mild forms of TRALI may pass unidentified.
- TRALI diagnosis in patients with other ALI risk factors, requires expert clinical evaluation, and still there are indeterminate cases.
- The definition does not include lab tests, so the diagnosis is only based on clinical variables.
- The limit of 6h cannot lead to detect cases developed after this period.
Recently, Mark & Corwin [7], have proposed an extension of the definition of TRALI in terms of the period in which the clinical manifestations begin: classic TRALI, which starts during the first 6 hours of transfusion and agrees with the model of immune TRALI, and late TRALI or TRALI-deferred, starting between 6 and 72h of transfusion and whose mechanism of production would poorly defined mediators.
2. PATHOPHYSIOLOGY OF TRALI
Pathophysiology of TRALI is not fully understood at present. In the 1950s, initial reports were made on the role of antileukocyte antibodies in the pathogenesis of TRALI, when it was administered to a test subject, 50 ml of blood from a patient containing leukoagglutinins. The clinical manifestations following this administration included hypotension, fever, respiratory failure and bilateral pulmonary infiltrates on chest, radiography with full recovery within 3 days.
There are two mechanisms, which lead to the development of the syndrome: immune-mediated and no immune- mediated TRALI.
i) Immune-mediated TRALI is caused by anti-HLA (human leukocyte antigen) antibodies class I, II and/or less frequent antibodies directed against specific antigens of granulocytes — HNA (human neutrophil antigen), which can be present in the serum of the recipient or donor, and react with the donor’s or recipient’s leukocytes, respectively [8, 9].
The Immune-mediated TRALI is estimated at 65-85% of all reported TRALI cases. Of antibody with specificity known, those directed against HNA-1a, HNA-1b, HNA-2a, HNA-3a (5b) and HLA-A2 are the most frequently documented [10]. Moreover, the presence of antibodies directed against HLA class II has been associated with TRALI.
An argument against immune model is that some patients who underwent the transfusion procedure did not develop TRALI, despite the presence of leukocyte antigens. Some arguments that can explain this issue are: i) heterozygosity antigen receptor recognized by the antibody; ii) the clinical presentation of transfused patient may predispose to a greater or lesser manifestation of pulmonary transfusion reaction, and iii) lack of detection by clinicians and lack of diagnosis of mild cases [11].
ii) Non-immune-mediated TRALI can be attributed to transfusion of biologically active compounds, which are accumulated in stored blood components such as bioactive lipids, proinflammatory cytokines or platelet microparticles with high procoagulant activity. In non-immune-mediated TRALI, no antibodies were detected despite the clinical symptoms [8].
The main inflammatory cells involved in TRALI pathogenesis are the polymorphonuclear neutrophils. For TRALI development, the production of leukocyte antibodies is crucial. There is a hypothesis of a “two-hit” model postulated by Silliman et al. 1995 [12], which assumes that TRALI onset depends on the coexistence of a factor that predisposes a recipient’s reaction, as well as on the presence of leukocyte-activating compounds in transfused blood.
The “first hit” is mediated by any proinflammatory condition, it is an aggression that activates the pulmonary endothelium and promotes the recruitment and adhesion of neutrophils to the capillary endothelium, and later causes a disarray ofthe lung alveolar-capillary permeability barrier [13].
The “second hit” is induced by transfusion of a blood component. These compounds induce neutrophils activation and release of cytotoxic factors that cause endothelial damage and capillary injury. Reactive neutrophils secreteproinflammatory mediators such as cytokines (IL-1, IL-6, IL-8, TNF-alpha), release proteolytic enzymes (e.g. elastase, azurocidin), and produce reactive oxygen species [8, 12]. These events initiate the cascade of immune reactions with the damage to the vascular endothelium, and produce pulmonary capillary fluid leakage in to the alveolar space, thus causing edema. The neutrophilic airway inflammation of the lung, and later the disruption of the lung alveolar-capillary permeability barrier, it seems to be similar to what is seen in other forms of ALI/ARDS, but with different origin [14].
There is a common pathway in the pathophysiology of TRALI, the damage to the alveolar-capillary membrane, where the neutrophil has been postulated as the protagonist cell for all reactions, and subsequently trigger as described above. Antibodies present in the transfused blood agglutinate to the first found neutrophils after transfusion, which are trapped in the microvasculature. It is well known that most of leukocyte antibodies of the IgG class, produce active neutrophils agglutination rather than passive. Neutrophils stimulated by leukocyte antibodies or biologically active lipids, liberate oxygen radicals which damage endothelial cells of the lung capillaries, leading to increased vascular permeability, and ultimately the passage of fluid and proteins into the alveoli [15].
The main pathophysiological events associated with TRALI are shown in Fig. (1).
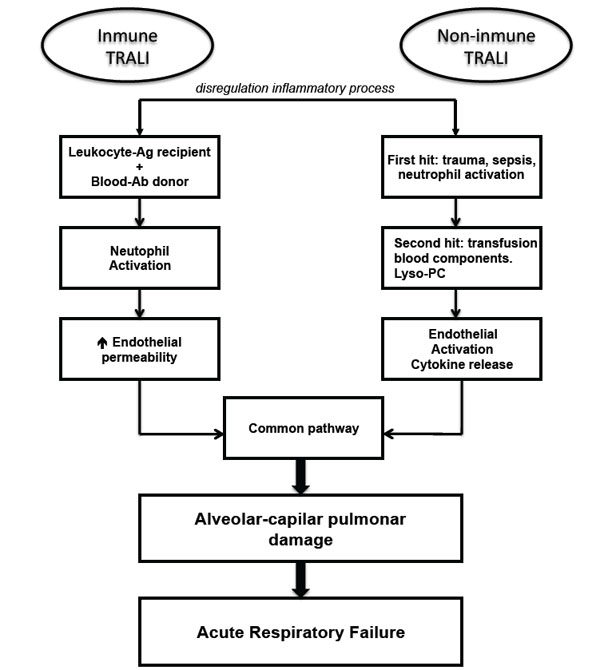
Pathophysiology of TRALI. Ag, antigen; Ab, antibody; Lyso-PC, Lysophosphatidylcholines.
3. EXPERIMENTAL MODELS
Animal models have disproved the microaggregate theory of acute lung injury from blood transfusions. The two major hypotheses of TRALI, passively transfused neutrophil and human leukocyte antigen antibodies, and biologically active lipids that accumulate in older, cellular blood products, have been replicated in animal models. In TRALI-related events, the transfused products contain a stimulus that is insufficient to cause apulmonary transfusion reaction in the absence of one or more separate facilitating recipient factors, which can synergize with the transfused factors.
3.1. Ex Vivo Lung Models of TRALI
In this type of TRALI models, edema is produced by perfusate containing human neutrophils, human anti-granulocyte alloantibody (anti-HNA 5b), and complement [16]. The lung edema resulted from an increase in vascular permeability and lung weight, which did not occur if the neutrophils of the perfusate were HNA 5b-negative or if any of the three components perfused was omitted. The HNA antibodies were also capable of direct neutrophil activation and reactive species oxygen (ROS) generation. Complement was not used, and the authors concluded that their model involves complement-independent antibody-induced neutrophil activation [17]. These data indicate that, direct antibody-mediated activation of cognate antigen expressing neutrophils may be largely responsible for lung injury due to anti-HNA antibodies, but provide contradictory evidence regarding the role of complement. Other investigators used rat ex vivo lung explants to model the non-antibody-mediated TRALI [18]. They administered a nonlethal injection of LPS to the animals, followed by lung isolation and perfusion with plasma from either fresh or stored blood products. LPS injection and plasma perfusion constituted the first and second event, respectively. TRALI-like changes (pulmonary artery pressure, edema, mediator-release, ALI-histology) were observed with plasma from stored packed red blood cells or platelets, but not with fresh plasma [19]. In this model, the LPS-preprimed activity of endogenous neutrophils, wich was already present in the explanted lungs was key for inducing TRALI changes. On the other hand, the antibody-mediated the ex vivo lung model [20], the monoclonal anti-HNA-2a antibodies are capable of direct activation of the HNA-2a-expressing neutrophils.
3.2. In Vivo Models of TRALI
These models are scarce in the literature, since there is a high variability in the pathophysiological responses of animals. Indeed, considerable investment in “trial and error” - based experimental approaches, may be needed in order to tease out the interacting variables that can produce TRALI-like reactions [21]. Some experimental variables are relevant in selected conditions for In vivo TRALI models: i) pharmacokinetic variables associated with TRALI-inducing agent, ii) induction of a “primed” state in the recipient, and iii) pathophysiological variables used for monitoring TRALI-events. The principal limitation of the developed TRALI models is related with pharmacological differences between the animals being used animals such as mice or rats, and the human physiology. In this case, the differential expression of leukocyte antigens has been described in rats and mice with TRALI or ARDS induced models [22].
The techniques for the development of ALI/ARDS in animal models, should also be applicable in modeling TRALI models. In particular, ALI/ARDS have been the central role of: i) activated neuthrophis, ii) activated pulmonary endothelium, iii) cell adhesion molecules on both these cells, as well as iv) a large amount of cytokines, v) other inflammatory mediators and vi) ROS [23].
4. CLINICAL PRESENTATION
The acute respiratory failure is the main finding in TRALI. The central clinical symptomis dyspnea and co-occurs with tachypnea, tachycardia, cyanosis and pulmonary secretions. Another hemodynamic alterations like fever, hypotension or hypertension (rare) are also reported [24]. All these clinical characteristics are observed during transfusion, or within 6 h of transfusion in the absence of other factors that can develop ALI, often during the first hour of a transfusion. During physical examination, the clinicians can find decreased breath sound, diffuse crackles over the lung fields, and shallow breathing. The chest radiography shows bilateral fluffy infiltrations with no cardiomegaly, and with this it can exclude cardiogenic edema. Hemodynamic monitoring helps to differentiate TRALI and cardiogenic pulmonary edema. Normal or low pulmonary capillary wedge pressure, and central venous pressure values are characteristic for TRALI [12, 25].
Actually, hemodynamic monitoring has been needed to exclude other causes of TRALI, because the symptoms are similar to other diseases, such as anaphylactic reaction, cardiogenic pulmonary edema, ARDS or bacterial infection, occasionally leukopenia, neutropenia, hypoalbuminemia and hypocomplementemia can be detected. The most prevalent symptom is a transient leukopenia, which arises in 5-35% of patients [26].
5. TREATMENT
The treatment of TRALI is the support of lung function, and it does not differ from the treatment of ALI/ARDS of any other etiologies. Mild cases of ALI/ARDS could be treated with conventional measures such as oxygen therapy, and more serious cases require endotracheal intubation and mechanical ventilation strategies. The protective strategies should be applied in ALI/ ARDS, which aim to prevent the damage produced by mechanical lung ventilation [27, 28].
Sometimes, the presence of hemodynamic impairment requires the administration of fluids. Aggressive adminis-tration is not recommended, since as in the ALI/ARDS of other etiology, it has recently confirmed that a conservative strategy is accompanied by an improvement in pulmonary function with decreased in duration of mechanical ventilation and the ICU stay [28]. The lack of response to fluid therapy may force the introduction introducing of the inotropes. The use of corticosteroids has been stated in some case reports, but their use in TRALI is empirical and there is no evidence of their use [29].
CONCLUDING REMARKS
TRALI is a distinctive clinical entity or a syndrome of ALI/ARDS, it has increased in clinical importance in recent years. Long unrecognized, and existing as only scattered reports of non-cardiogenic pulmonary edema after transfusions, TRALI remained largely undiagnosed even after a case-series report. The relative contribution of biological response modifiers, and anti-leucocyte antibodies to clinical TRALI remains unclear, and it may be difficult to answer using animals and in vitro models. It seems likely that (i) traditional molecular mediators of inflammation are among such molecules critical for the manifestation of TRALI; (ii) the action of such mediators can be inhibited by appropriate antagonists; and (iii) results can be translated into therapeutic strategies for clinical use. The identification of risk factors further improves the risk-benefit assessment of a blood transfusion. Efforts to further decrease the risk of TRALI are needed to increase awareness of this syndrome among physicians.
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest.
ACKNOWLEDGEMENTS
Author’s Role: All authors helped to write the manuscript and have seen and approved the final version.

