All published articles of this journal are available on ScienceDirect.

Changes in Clinical Markers During A Short-Term Transfer Program of Adult Cystic Fibrosis Patients from Pediatric to Adult Care
Authors Info & Affiliations
Abstract
Background:
Transition from child-oriented to adult-oriented health care in Cystic Fibrosis (CF) has become more important over recent decades as the survival of people with this disease has increased. The transition process usually begins in adolescence, with full transfer completed in early adulthood.
Objective:
This study investigated the impact of a short-term transfer program on clinical markers in an adult CF cohort still being managed by pediatricians.
Methods:
Clinically relevant data from the year before (T-1), the time of Transfer (T) and the year after the transfer (T+1) were analysed retrospectively.
Results:
39 patients (median age 29.0 years; 64% male) were transferred between February and December 2016. Lung function had declined significantly in the year before transfer (in % predicted: Forced Expiratory Volume in 1 second (FEV), 62.8 vs. 57.7, p <0.05; Forced Vital Capacity (FVC), 79.9 vs. 71.1, p<0.05), but remained stable in the year after transfer (in % predicted: FEV: 56.3; FVC 68.2). BMI was stable over the whole observational period. There was no relevant change in chronic lung infection with P. aeruginosa, Methicillin-Resistant Staphylococcus aureus (MRSA) and Burkholderia sp. during the observation period. The number of patient contacts increased significantly in the year after versus the year before transfer (inpatient: 1.51 vs. 2.51, p<0.05; outpatient: 2.67 vs. 3.41, p<0.05).
Conclusions:
Our data show that, within the framework of a structured transfer process, it is possible to transfer a large number of adult CF patients, outside a classic transition program, from a pediatric to an adult CF center in a short period of time, without any relevant changes in clinical markers and, stability.
1. INTRODUCTION
The transition of chronically ill children from child- to adult-orientated health care should be conducted within the framework of a structured transition program, starting in adolescence [1, 2]. Transition describes “a purposeful, planned process that addresses the medical, psychosocial and educational/ vocational needs of adolescents and young adults with chronic physical and medical conditions as they move from child-centred to adult-oriented health systems” [3]. Especially in Cystic Fibrosis (CF), the most common autosomal-recessive disorder in Caucasians, the process of transition has gained increasing attention as life expectancy in CF patients increases continuously. The implementation of newborn screening programs [4], improved treatment paradigms, and the development of new drugs [5] led to an increase in average life expectancy for CF patients in the Western countries to over 40 years of age [6]. Therefore, CF is no longer a pediatric but also an adult disease [7, 8]. Registry data indicate that the number of adult CF patients will increase even more within the next years [9]. Consequently, CF-associated comorbidities of adulthood have become more important and require specialised care [10]. However, given that CF care used to be a pediatric domain in the past, many adult CF patients remain under pediatric care [11, 12].
Recently, detailed descriptions of transition programs for CF patients were published [13-18]. None of these has proven to be superior to the others [19]. Expert committees recommend an early start of the transition process involving patients, their parents, and families [20, 21]. Existing programs focus on adolescents and young adults and deal with concerns about the transfer, transition readiness, psychosocial needs, and variations in care [22]. The transition into adulthood is obviously challenging for most adolescents and even more complex for those with chronic diseases. Pediatric care tends to be often very protecting, whereas adult care is supposed to be more self-directed. In addition, transition usually takes place during a vulnerable phase for CF patients in which we frequently observe a decline in lung function, a decrease in Body Mass Index (BMI), an increased rate of hospitalisations, and developmental and psychosocial changes [23]. Adolescents or young adults with CF often have a long history with their pediatric CF center, resulting in strong emotional bonds to and trustful relationships with their CF care team [24, 25]. A change in the CF care team potentially leads to in a different organising structure as well as a shift of perspective, treatment approach, interviewing, and knowledge. Overprotection, meaning strong support of pediatric care may harm the transition process; a mechanistic and pharmacotherapy oriented treatment approach in the adult units might overburden those overprotected CF families. Most transition concepts deal with adolescents and their parents and how the transition to adult medicine can be guaranteed without getting lost in the adult world. Although most adult CF patients have access to adult pulmonologists, it is not uncommon for adult patients in many European countries to be treated by pediatricians in pediatric wards [26]. Due to structural changes at the children´s hospital we needed a program to transfer a large group of adult CF patients to an appropriate adult-based care in less than one year.
The current analysis was conducted to determine the impact of a short-term transfer program on CF-relevant clinical data, such as lung function, BMI, number of in- and outpatient contacts, change in bacterial and fungal colonisation and serious clinical events (e.g., pneumothorax, severe haemoptysis, and lung transplantation). Especially in Germany, a significant number of CF patients remain in pediatric care. Patients and centers probably profit from a structured short-term transfer program as described here.
2. METHODS
2.1. Study Design
This retrospective analysis included all patients transferred from the Cystic Fibrosis Center of the Pediatric Pulmonology at the University Children´s Hospital Essen to the Adult Cystic Fibrosis Center at the Ruhrlandklinik Essen between February and December 2016. The study was approved by the ethics committee of the University Hospital Essen (17-7854-BO).
2.2. Patients
A total of 43 CF patients > 18 years of age who were receiving care at the pediatric clinic in mid-2015 were considered for participation in the transfer program.
2.3. Transfer
The short-termtransfer followed astructured process (Fig. 1). In summer 2015, preparation for the upcoming transfer process was started. First, all eligible patients were identified and received a comprehensive elucidation about the need for a transfer from pediatric to adult care during a regular outpatient visit or inpatient stay. The transfer process was described explicitly by the treating pediatrician, the CF nurses, and the psychosocial workers. The first outpatient visit at the adult CF center was scheduled by the attending physicians. Internal CF team meetings were conducted to discuss all relevant medical and psychosocial issues concerning the patients prior to the first patient contact. On the first presentation at the adult center, each CF patient was seen by both, a CF-specialist from the Pediatric and the Adult Cystic Fibrosis Center. Parents, relatives, and friends were invited to attend the first meeting. This was, again, to guarantee a thorough exchange of information between the previously treating physicians, the new doctors, and the patient. Following the doctor-patient talk, the first contact with other parts of the CF team was initiated. In conversation with representatives from nutritional counselling, psychological and psychosocial services, patients had the chance once more to focus on their individual situation and to discuss outline current problems. As a part of these discussions, a comprehensive overview of the procedures and structures of the new clinic was presented to the patient. Finally, we explained contact options to ensure outpatient medical care and how to deal with emergency situations.
The aim of our practice was to maximise patient acceptance of the process and to perform a comprehensive initial assessment. To complete the transfer, all patient paper-based charts were copied and transferred from the pediatric to the adult center. Radiological findings were provided electronically.
The data collected by each member of the CF team were recorded. In regular team meetings, the collected data were discussed in order to prevent a loss of information and to identify individual problems of our patients.
2.4. Assessments
The first in- or outpatient visit at the adult CF unit was defined as the time of transfer (T0). Times T-1 and T+1 refer to 1 year before and after the transfer, respectively. Lung function, bacterial colonisation, nutritional status and out- and inpatient contacts were determined from patient medical records, and occurrence of relevant clinical events (lung transplantation, pneumothorax, severe haemoptysis, pregnancy, and death) was identified. Paper and electronic-based medical records were also used to identify the presence of comorbid conditions (e.g., diabetes mellitus, liver impairment/cirrhosis, allergic bronchopulmonary aspergillosis, and pregnancy/ paternity), and the use of any supportive therapy (e.g., oxygen therapy, non-invasive ventilation).
All regular (every three months) or unscheduled contacts to the pediatric or adult CF center included microbiologic surveillance (sputum culture or throat swab), calculation of the BMI and lung function testing. Forced vital capacity (FVC) and forced expiratory volume in 1 second (FEV1) were measured in both clinics with a JAEGER MasterScreen Body (CareFusion, Hoechberg, Germany) according to the ATS guidelines [27].
2.5. Statistical Analysis
Statistical analysis was performed using version 22 of the SPSS statistics package (SPSS Inc., Chicago, USA). The figures were plotted using Prism (version 6.05; GraphPad, Inc., San Diego, CA). Data are presented as mean, standard deviation, and range. The Kolmogorov-Smirnov test was used to test for normal data distribution. Comparisons between groups were performed using the paired t-test or Wilcoxon test. Correlations between FEV1 course and contacts to the CF centers were performed using Spearman correlation.
3. RESULTS
3.1. Patients
In all 43 patients who were eligible for transfer, CF was diagnosed by molecular genetic testing. 39 out of 43 patients (mean age 29.0 ± 9.3 years) were actually transferred to the Adult Cystic Fibrosis Center at the Ruhrlandklinik Essen and were included in this retrospective analysis (Table 1). Three patients presented independently at the adult clinic without participation in the structured transfer process, and one was transferred to another CF center closer to his home.
3.2. Lung Function and BMI
All the patients had reduced lung function (Table 2). FEV1 (Fig. 2) and FVC (Fig. 3) significantly declined in the year prior to transfer, did not change during the transfer process, and then remained stable in the year after transfer. BMI did not change significantlyduring the whole transfer process (Table 2).
Results are presented as mean ± standard deviation, * p<0.05. T, transfer; T-1, year before transfer; T+1 = year after transfer.
Results are presented as mean ± standard deviation, * p<0.05. T, transfer; T-1, year before transfer; T+1 = year after transfer.

| Patient Characteristics | Subjects (n=39) |
|---|---|
| Age, years | 29.0 ± 9.3 (18–57) |
| Male, n (%) | 25/39 (64) |
| Genotype | |
| Δ F508 homozygous, n (%) | 22 (56) |
| Δ F508 heterozygous, n (%) | 14 (36) |
| Other, n (%) | 3 (8) |
| BMI, kg/m2 | 20.9 ± 3.2 (16.8–30.1) |
| FEV1, L | 2.2 ± 1.1 (0.82–4.49) |
| FEV1, % predicted | 57.7 ± 27.0 (24–120) |
| FVC, L | 3.2 ± 1.3 (1.05–5.53) |
| FVC, % predicted | 71.1 ± 25.6 (29–135) |
| Pancreatic insufficiency, n (%) | 35/39 (90) |
| Pseudomonas aeruginosa positive, n (%) | 21/39 (54) |
| Cystic fibrosis-related diabetes, n (%) | 11/39 (28) |
BMI, Body Mass Index; FEV1, Forced Expiratory Volume in 1 second; FVC, Forced Vital Capacity.
| Subjects (n=39) | T-1 (–1 year) | T0 (Transfer) | T+1 (+1 year) |
|---|---|---|---|
| BMI, kg/m2 | 21.0 ± 3.24 (14.2–29.8) | 20.9 ± 3.21 (15.4–30.1) | 21.2 ± 3.59 (14.2–30.8) |
| FEV1, L | 2.36 ± 1.20 (0.78–4.54) | 2.18 ± 1.11 (0.82–4.49)a | 2.22 ± 1.14 (0.74–4.88) |
| FEV1, % predicted | 62.8 ± 29.9 (23–119) | 57.7 ± 27.0 (24–120)a | 56.3 ± 25.6 (21–111) |
| FVC, L | 3.52 ± 1.25 (1.32–6.11) | 3.16 ± 1.31 (1.05–5.53)a | 3.23 ± 1.36 (1.35–5.76) |
| FVC, % predicted | 79.9 ± 25.5 (34–138) | 71.1 ± 25.6 (29–135)a | 68.2 ± 23.1 (27–123) |
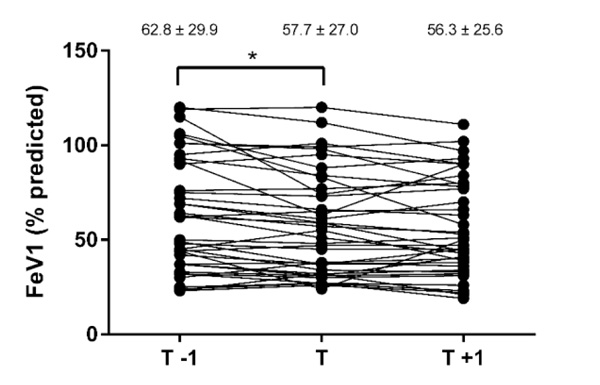

3.3. CF Center Contacts
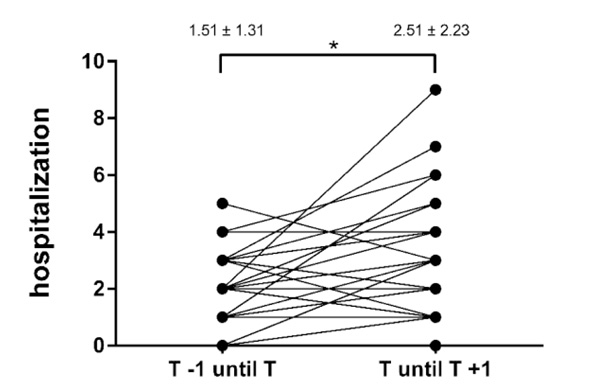
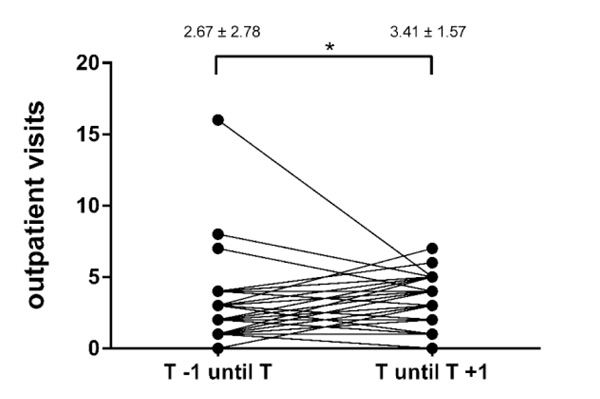
The frequency of both in- and outpatient visits significantly increased during the first year after the transfer (Table 3, Figs. 4 and 5). There was no significant association between lung function decline and cumulative contacts to the CF centers in the year before (r =0.05, p =0.76) or after (r = –0.008, p =0.96) patient transfer.
Results are presented as mean ± standard deviation, * p<0.05. T, transfer; T-1, year before transfer; T+1 = year after transfer.
Results are presented as mean ± standard deviation, * p<0.05. T, transfer; T-1, year before transfer; T+1 = year after transfer.
| Subjects (n=39) | T-1 to Transfer | Transfer to T+1 | p-value |
|---|---|---|---|
| Hospitalization | 1.51 ± 1.31 (0–5) | 2.51 ± 2.23 (0–9) | <0.05 |
| Outpatient visits | 2.67 ± 2.78 (0–16) | 3.41 ± 1.57 (0–7) | <0.05 |
| Cumulative contacts | 4.18 ± 3.2 (1–19) | 5.92 ± 2.22 (2–11) | <0.05 |
| Subjects (n=39) | T | T+1 |
|---|---|---|
| Pseudomonas aeruginosa, n (%) | 21 (53.8) | 21 (53.8) |
| 3/4-MRGN Pseudomonas aeruginosa, n (%) | 13 (33.3) | 14 (35.9) |
| MSSA, n (%) | 15 (38.5) | 20 (51.3) |
| MRSA, n (%) | 4 (10.3) | 4 (10.3) |
| Burkholderia sp., n (%) | 2 (5.1) | 2 (5.1) |
| Stenotrophomonas maltophilia, n (%) | 1 (2.6) | 2 (5.1) |
| Achromobacter sp., n (%) | 7 (17.9) | 7 (17.9) |
| Aspergillus fum., n (%) | 15 (38.5) | 16 (41.0) |
3.4. Changes in Microbial Colonisation
There were no relevant changes in microbial colonisation during the entire transfer period. The majority of the patients were chronically infected with P. aeruginosa (53.8%), which was classified as multi-drug-resistant in more than half of the infected patients. The number of chronic P. aeruginosa, methicillin-resistantStaphylococcus aureus, or Burkholderiasp. infections remained constant during the whole observation period (Table 4).
3.5. Clinical Events
In the first year after the transfer, 5/39 (13%) patients had relevant clinical events. Two patients underwent lung transplantation in the first year after transfer to the adult clinic due to significant clinical deterioration. Both patients suffered from ventilatory insufficiency with the need for establishing non-invasive ventilation therapy, which led us to list those patients for lung transplantation. One of the lung transplant recipients died shortly after transplantation due to right heart failure. One patient had significant haemoptysis that needed to be treated with a bronchial arterial occlusion procedure. One female patient became pregnant and gave birth to a healthy child. And in one patient, a spontaneous pneumothorax could not be treated adequately by application of a chest tube which led to surgical intervention; no recurrence was recorded.
4. DISCUSSION
To our knowledge, this analysis is the first description of a short-term transfer program of a large adult CF cohort from a pediatric to an adult CF center.
Often, pediatric CF centers need to implement a short-term transition program because adult cohorts continue to occupy pediatric capacities in large tertiary hospitals where those units are usually located [28]. The first step to achieve a successful transfer is to implement a structured transition process, which is time-consuming, requires sufficient staff, and extends over many years.
With the increasing life expectancy of CF patients, special medical care structures are needed to meet the needs of aging patients [29], which are usually not covered by pediatric CF units. Many pediatric CF centers realise that they do not fulfil all the needs of adult CF patients. Professional societies recommend an early start of the transition process, involving parents and relatives, and close cooperation between the participating institutions, including the caregiving teams [30]. However, in practice, many pediatric CF centers throughout Europe are located at children's hospitals. Those centers care for a large number of adult CF patients but have failed to implement a transition program in time, which is to be started in adolescence [26]. This is why a short-term transfer program is of interest for many CF centers.
The two CF centers in Essen referred to in his study have been working together closely for years. Therefore, we could easily set up a structured short-term transfer program. After identification of all possible transfer candidates by the pediatricians, an appointment for each patient was made in the adult CF center. At this first appointment, two experienced CF practitioners met with the patient in an outpatient setting. This appointment included the structured transfer of the patient from the pediatric to the adult clinic and the introduction of the medical and caregiver teams.
Although CF is now known to be an adult disease, awareness of CF in adult pulmonary medicine is generally limited [31]. In Germany in particular, this results in a lack of specialised adult CF centers. The few established centers are usually large and have a rapidly growing number of adult CF patients and quickly reach their capacity limit. Both pediatric and adult CF centers need to change their practice; e.g., pediatric CF centers need to relinquish care of adult CF patients, and adult centers need to increase their capacity to maintain specialized medical care.
In our cohort, the mean age at the time of transition was 29.0±9.3 years, which is significantly older than in all published studies investigating clinical outcomes after transition [32]. Mean age-adjusted FEV1 and FVC values in our adult CF-cohort at the time of transfer were comparable to those in the US CF registry [33]. Our analysis showed that FEV1 and FVC decreased from timepoint T-1 to T, but stabilised in the year after transfer.
Previously published data also showed stable lung function after a structured transition [32-34].
At time T, lung function was first performed in the adult clinic. The Global Lung Initiative (GLI) values were used as reference values. In contrast, the previous measurements of pulmonary function were performed in the pediatric clinic using Zapletal values as reference. These values are not fully comparable because GLI values are lower than Zapletal values [35]. This appears as a reason for the decline in lung function from T-1 to T. This also indicates that between the times T and T + 1, no significant difference in lung function can be measured.
In our patients, body weight and BMI remained stable over the 2-year observation period, consistent with data from other established transition programs [36]. There were no relevant changes in the microbiology of sputum cultures in the year after the transfer, suggesting that the transfer does not increase the risk to acquire new pulmonary infections.
In contrast to other studies [34], we observed an increase in outpatient and inpatient contacts in the first year after transfer. There are several explanations for that: one possible reason is the significantly younger age of the evaluated patients in previously published studies with a reduced likelihood of disease-related complications. As the patient’s age increases and lung function worsens, the rate of complications increases [37-39]. In the presented cohort, the rate of major clinical events was very high (5/39 patients, 13%). Due to the occurrence of relevant clinical events, in some patients, closer patient monitoring and intensified therapy were necessary in the year after transfer,which inevitably led to an increase in doctor-patient contacts. Another reason for the increase of the center contacts after the transfer is that eight of the transferred patients were included in clinical trials exclusively open to adult CF patients. Patients in clinical trials wereseen additionally on study visits resulting in closer medical supervision. So, the increase in doctor-patient-contacts was not to be seen as a marker for disease worsening only. Participation in clinical trials improved adherence to medical treatment [40]. This close and comprehensive care can help remove patient barriers leading to better compliance with and adherence to CF therapy, which in turn has a positive effect on disease process [41].
Nevertheless, keeping a critical eye on ourselves, the relatively short assimilation phase for patients in the adult clinic also played a role. The longer patients and therapists know each other, the easier medical problems of the individual can be assessed. After all, the large number of contacts within the first year after the transfer is at least partially a result of the brief introduction phase. The analysis presents results from a retrospective study, with partially incomplete data sets. There was no systematic evaluation of the transfer process from the patients’ point of view, e.g., by using questionnaires to assess the impact of health-related quality of life before and after transfer. In addition to CF relevant clinical data documented by us, this additional information could be obtained in the future in order to further optimize the transfer process.
CONCLUSION
Our data show that a short-term transfer program of meanwhile adult CF patients, based on excellent interdisciplinary collaboration between all professional groups who are dedicated to the care of CF patients, facilitates a successful transfer of a large number of patients from a pediatric to an adult CF center in a very short time. This procedure, even without an established transition program, is safe for patients and does not lead to relevant changes in clinical outcome parameters important for CF.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study was approved by the ethics committee of the University Hospital Essen (17-7854-BO).
HUMAN AND ANIMAL RIGHTS
No Animals were used in this research. All human research procedures followed were in accordance with the ethical standards of the committee responsible for human experimentation (institutional and national), and with the Helsinki Declaration of 1975, as revised in 2013.
CONSENT FOR PUBLICATION
Informed consent was obtained from all the participants prior to publication.
FUNDING
None.
CONFLICTS OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
AUTHORS' CONTRIBUTIONS
Conception, Data collection and study design: MW, SS, CT, MO, UM, FS
Manuscript preparation: MW, FS
Analysis and interpretation: MW, FS
Review of the manuscript for important intellectual content: MW, SS, CT, MO, UM, FS
ACKNOWLEDGEMENTS
The authors thank all families that participated in this study, and Manuela Groch-Seidler, Ramona Hellwig and Nadine Kordt for technical assistance.

