All published articles of this journal are available on ScienceDirect.

Caveolin-1 Expression and Hemodynamics in COPD Patients
Authors Info & Affiliations
Abstract
Caveolin-1 is a regulator of both intracellular calcium homeostasis and endothelial nitric oxide synthase and may play a pathogenetic role in pulmonary hypertension. In the present study, we aimed to investigate the correlations between pulmonary hemodynamics and vessel morphology including the expression of Caveolin-1 in pulmonary arterioles from patients with chronic obstructive pulmonary disease (COPD) who underwent lung-volume reduction surgery. Staining and subsequent analysis was performed on paraffin-embedded lung tissue from COPD patients (n = 12). Pulmonary arteries with an external diameter of 100-500µm were analysed. Immunhistochemistry with antibodies against caveolin-1 was performed and intensity was assessed. Morphometric data were obtained by using computer-assisted imaging software. The findings were quantified and correlated to hemodynamic data obtained by right-heart catheterization. In COPD patients with pulmonary hypertension (n = 5), the expression of caveolin-1 within the medial smooth muscle cell layer was found to be increased, whereas the intimal caveolin-1 was more prominently expressed in COPD patients with normal pulmonary pressures (n = 7). The ratio between these expression patterns was positively correlated to the mean pulmonary artery pressure. Similar findings were observed for the ratio between intimal and medial thickness as well as for the expression of smooth muscle actin (SMA).
Taken together, the expression of caveolin-1 within medial smooth muscle cells of pulmonary arteries in patients with COPD is associated with pulmonary hypertension. Our results thus emphasize a potential novel player in the pathogenesis of COPD-associated pulmonary hypertension.
BACKGROUND
Chronic obstructive pulmonary disease (COPD) is the fourth leading cause of death in industrialized countries [1]. One factor contributing to the devastating outcome of end-stage COPD is the development of pulmonary hypertension and cor pulmonale. In cross-sectional studies, pulmonary hypertension and right heart failure have been found in more than 10% of patients dying from COPD [2, 3]. The arterial vessels in idiopathic pulmonary hypertension are characterized by intense thickening of their walls through fibromuscular proliferation and microthrombotic occlusion [4, 5], whereas in the setting of COPD, vasoconstriction induced by hypoxia may play a pivotal role. In either case, the molecular factors mediating the vascular changes that increase pulmonary vascular resistance in COPD patients remain largely unravelled so far.
Recently, the role of caveolins (Cav) as emerging signalling molecule has been investigated in animal models of pulmonary hypertension as well as in lung tissue samples from patients with idiopathic pulmonary arterial hypertension [6-9]. Cav-1 is a 22kDa protein located in cholesterol- and glycosphingolipid enriched microdomains (lipid rafts or caveolae) that act as chaperons and scaffolding regions in order to anchor and regulate proteins [7, 10, 11]. In this regard, Cav-1 is involved in the regulation of many cellular processes including signal transduction and calcium homeostasis [12]. Moreover, a strong relationship between Cav-1 and endothelial nitric oxide-synthase (eNOS) has been described (reviewed in [13]) and binding of eNOS to Cav-1 appears to inhibit the enzymatic function [14]. Both, changes in the intracellular calcium-storage resulting in increased vascular tone and a reduction in the production and release of endothelial NO, have been associated with the pathogenesis of pulmonary hypertension. The knockout of the cav-1 gene in mice resulted in lung fibrosis and subsequent development of cor pulmonale [9, 15]. Conversely, a recent study by Patel has found an increased expression of Cav-1 in smooth muscle but not in endothelial cells in pulmonary arteries from patients with idiopathic pulmonary arterial hypertension [7].
Along this line, we investigated the vessel morphology and expression of Cav-1 in small pulmonary arteries from COPD patients that underwent lung-volume reduction surgery (LVRS).
METHODS AND MATERIALS
Patients/Tissue Sections
Paraffin-embedded lung tissue sections were obtained from COPD patients undergoing lung volume reduction surgery or lung transplantation. Pulmonary hemodynamics was assessed by right-heart catheterization performed before surgery. Patients with a mean pulmonary arterial pressure (mPAP) ≤ 20mmHg were chosen as controls (n = 7), whereas patients with a mPAP ≥ 25mmHg were considered to have pulmonary hypertension (n = 5). The experimental use of these data was approved by an internal review board of the local Ethics committee of Zurich. In addition, all patients gave written informed consent for further experimental analysis of their explanted tissue and clinical data. Details of patients’ characteristics are provided in Table 1.
Characteristics of Analysed Patients
| COPD with PH (n = 5) | COPD without PH (n = 7) | |||
|---|---|---|---|---|
| age | 56 ± 11.9 [42-72] | 67 ± 9.0 [54-79] | ||
|
|
||||
| gender (m: f) | 2: 3 | 4: 3 | ||
|
|
||||
| ethnic group | Caucasian | Caucasian | ||
|
|
||||
| mPAP (mmHg) | 29.6 ± 5.1 [38-26] | 16.7 ± 2.7 [20-12] | ||
|
|
||||
| spirometric data | ||||
| - FVC (% predicted) | 59 ± 19.8 | 59 ± 12.9 | ||
| - FEV1 (% predicted) | 26 ± 7.3 | 27 ± 5.8 | ||
|
|
||||
| SaO2 (%) | 89 ± 5.2 | 94 ± 3.9 | ||
|
|
||||
| smoking status | 5/5 | 7/7 | ||
| - pack years | 62 ± 53.1 [10-150] | 61 ± 22.3 [35-100] | ||
Abbreviations used: FVC forced vital capacity, FEV1 forced expiratory volume in one second, SaO2 oxygen saturation in arterial blood.
Vessel Morphometry
| COPD with PH (n = 5) | COPD without PH (n = 7) | |
|---|---|---|
| vessels analysed/ slide | 7 | 7 |
|
|
||
| diameter of pulmonary artery (µm)¶ | 223.1 ± 85.9 [110-419] | 292 ± 88 [125-465] |
|
|
||
| diameter of EEL (µm) ¶ | 193.6 ± 83.6 | 265.6 ± 80.6 |
|
|
||
| diameter of IEL (µm) ¶ | 169.8 ± 75.4 | 233.3 ± 76.6 |
|
|
||
| intimal area (mm2) | 10.0 ± 10.7 | 16.4 ± 13.5 |
| - % of total vessel area | 31.7 ± 16.3 | 40.8 ± 16.8 |
|
|
||
| media area (mm2) | 7.9 ± 6.9 | 13.1 ± 12.2 |
| - % of total vessel area | 27.3 ± 11.1 | 32.1 ± 17.6 |
|
|
||
| adventitial area (mm2) | 10.0 ± 6.6 | 12.6 ± 13.2 |
| - % of total vessel area | 40.4 ± 16.4 | 27 ± 19 |
|
|
||
| caveolin expression | ||
| - intimal caveolin | 2.34 ± 0.52 | 2.60 ± 0.37 |
| - medial caveolin | 1.78 ± 0.73 | 1.29 ± 0.5 |
¶ Narrowing index applied.
Theoretical diameter was calculated by dividing the measured perimeter by π.
EEL = external elastic layer.
IEL = internal elastic layer.
Immunohistochemistry
Serial sections of 2μm were manufactured from tissue blocks and were stained with hematoxylin eosin (HE) and Elastica van Gieson (EVG). Immunhistochemistry was performed on a Ventana automat (Ventana Medical Systems) with antibodies directed against Cav-1 (BD Transduction Laboratories, mouse monoclonal, clone 2297, 1: 100) and smooth muscle actin (α-SMA) prior to analysis by light microscopy. For analysis, staining was assessed on 3 vessels/slide in a semiquantitative manner by using a visual scoring system (0 = no staining, 1 = weak, 2 = moderate, 3 = strong).
Morphometry
Morphometry data were obtained by using computer-assisted imaging software (Axiovision, Zeiss). Pulmonary arteries with an external diameter of 100-500μm were randomly chosen followed by analysis (n = 7 per slide and individual patient). Only vessels with a cross-sectional morphology were chosen to minimize artificial effects by longitudinal or tangential cuts. Additionally, tissue shrinkage was corrected by calculation of a narrowing index. The adventitial layer, the external and internal elastic laminas (EEL and IEL, respectively) encompassing the vessel’s smooth muscle cell layer (medial layer) as well as the intimal layer were outlined manually at the screen. The perimeter and the corresponding diameter were calculated automatically by the imaging software. Moreover, the diameter was corrected by a narrowing index to avoid artificial vascular contraction due to tissue shrinkage [16]. Thus, the measured perimeter was divided by π to calculate a theoretical diameter of a fully extended vessel. Lumen as well as intimal, medial and adventitial layer areas were calculated and expressed as percentage of the whole vessel area. The final data per patient was expressed as mean of all vessels analysed.
Statistics
For statistical analysis, the unpaired t-test was used to compare data from the two groups. Correlation analyses (correlation coefficient Pearson r) were performed to assess correlations between pulmonary hemodynamics, thickness of vessel layers and immunohistochemical expression of distinct proteins. Statistics were performed using GraphPad Prism Software system (GraphPad Software, San Diego, CA, USA) and a p value <0.05 was considered to be statistically significant. All data are shown as mean ± SD.
RESULTS
Vessel Morphometry
As shown in Table 2, a total of 7 vessels were investigated for each individual patient with similar mean diameters of 223.1 ± 85.9 μm for COPD patients with pulmonary hypertension and of 292 ± 88 μm for COPD patients without pulmonary hypertension, respectively. The intimal layer area was found to be strongly reduced from 40.8 ± 16.8% in patients without pulmonary hypertension to 31.7 ± 16.3% in patients with pulmonary hypertension. No significant changes have been observed in the thickness of the smooth muscle cell area between the two groups. The thickness of the adventitial area on the other hand, was significantly increased in patients with pulmonary hypertension from 27 ± 19% to 40.4 ± 16.4%, respectively (p = 0.05).
When analysed by subsequent linear regression analysis (Fig. 1), the ratio between the intimal and the medial vessel layer was positively correlated to the mean pulmonary arterial pressure (r2 = 0.49). Similarly, the thickness of the adventitial layer expressed as percentage of the whole vessel was strongly correlated to the mPAP (r2 = 0.67).

Correlation analyses. (A) Parametric analyses to calculate the Pearson r correlation coefficient showed strong associations between pulmonary hemodynamics (mPAP) and the ratio between the thickness of medial and intimal vessel areas (r2 = 0.49). (B) Significant correlation were also found between mPAP and the ratio between medial : intimal Cav-1 expression (r2 = 0.53, p = 0.007). (C) A similar correlation was found between mPAP and the ratio between medial : intimal α-SMA expression (r2 = 0.48).
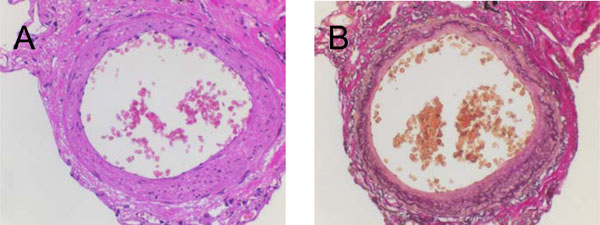
Cross-sectional imaging studies of a pulmonary artery (IHC). Serial stainings performed at lung tissue slides derived from an index patient without pulmonary hypertension: (A) Hematoxylin-Eosin (HE) and (B) Elastin-van Gieson (EVG).
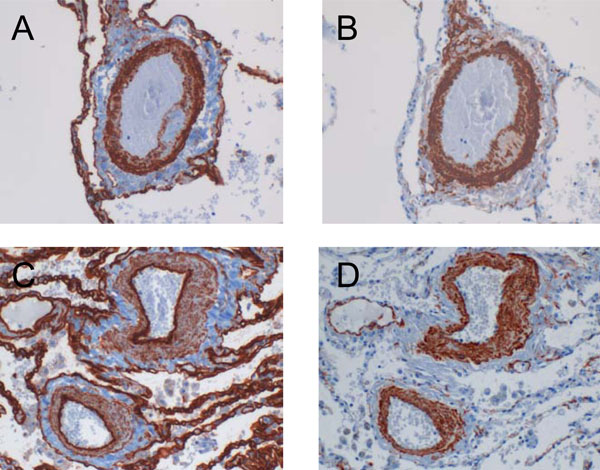
Cav-1 expression pattern in COPD patients with and without pulmonary hypertension. (A) Cav-1 was found strongly expressed within the media of pulmonary arteries derived from COPD patients with pulmonary hypertension as compared to their counterparts without pulmonary hypertension (C). Conversely, the intimal expression of Cav-1 was increased in patients without and decreased in patients with high pulmonary pressure. Serial stainings for α-SMA are shown in B and D, respectively.
Caveolin-1 Expression
By using a visual scoring system, immunohistochemical stainings of pulmonary arteries were assessed in a semiquantitative manner (Fig. 2). The expression of Cav-1 within the smooth muscle cell layer was found to be increased in COPD patients with pulmonary hypertension as compared to the individuals without pulmonary hypertension (1.78 ± 0.73 versus 1.29 ± 0.5). On the other hand, the intimal Cav-1 was most prominently expressed in COPD patients without pulmonary hypertension (2.6 ± 0.37), but appeared to be reduced in patients with pulmonary hypertension (2.34 ± 0.52). Fig. (3) shows representative images of the Cav-1 expression pattern in pulmonary arteries of COPD patients with and without pulmonary hypertension.
Subsequent correlation analysis found a strong positive association between the mPAP and the Cav-1 expression pattern when expressed as ratio between medial and intimal Cav-1 (r2 = 0.53, p = 0.007) as shown in Fig. (1b). A similar relationship was detected between the expression of α-SMA in the intimal and medial layer and the corresponding mPAP (Fig. 1c).
DISCUSSION
Caveolins have emerged as novel signalling molecules acting as signal transducers and mediators of calcium homeostasis. The isoform Cav-1 has been particularly investigated in animal models of pulmonary hypertension and, by one pilot study, in human tissue samples derived from patients with idiopathic pulmonary arterial hypertension [6-9].
Our investigation of the vessel morphometry and the Cav-1 expression in small pulmonary arteries derived from COPD patients with and without concomitant pulmonary hypertension showed a reduction of the Cav-1 expression in the intimal layer of the vessels in patients with pulmonary hypertension. Conversely, the Cav-1 expression in the smooth muscle cell layer was found to be increased in COPD patients with pulmonary hypertension. The ratio of either trend, assessed in a semiquantatitive manner at the light microscopy, was associated with rising pulmonary arterial pressure. Since similar changes were also observed in the expression pattern of an independent smooth muscle cell marker (i.e. α-SMA), it might be suggested that the Cav-1 expression is strongly linked to the vascular distribution of smooth muscle cells.
In pulmonary hypertension, the weak expression of Cav-1 in the intimal layer might be due to an accelerated loss of caveolar peptides through a disrupted and dysfunctional endothelium. Since the bone morphogenetic protein receptor type II (BMPR2), one of the pathogenetic key molecules involved in the development of pulmonary hypertension, co-localizes with caveolins [17], the loss of Cav-1 might also be explained by the downregulation of the BMPR2 at the endothelial surface of pulmonary arteries as it has been described repeatedly in pulmonary hypertension. The elevated vascular resistance in pulmonary hypertension is mediated by calcium ions that promote the contraction of smooth muscle cells on the molecular level. The weak expression of Cav-1 in the intimal area of pulmonary arteries thus could alternatively be explained by an enhanced transcytosis of calcium by [Ca2+]-influx channels and ion pumps located on caveolar domains through the endothelium to the smooth muscle cell layer. Conversely, the high expression of Cav-1 observed within the vascular smooth muscle cells in COPD patients with pulmonary hypertension is probably related to increased levels of sarcoplasmic [Ca2+] due to highly active transient receptor potential channels. It is unclear at the moment, whether the abundant expression of Cav-1 within vascular smooth muscle cells is directly connected to the upregulation of other important signalling molecules in COPD patients with pulmonary hypertension. However, several molecules with pathogenetic relevance for the development of pulmonary hypertension have been located on caveolins including epidermal growth factor (EGF), platelet-derived growth factor (PDGF), receptors for endothelin and serotonine transporters [8, 18-23]. Our data thus support the hypothesis that caveolins are involved in the regulation and expression of these factors and, in turn, are of pathogenetic importance for the development of pulmonary hypertension.
The clinical relevance of our findings, on the other hand, is less clear and has to be addressed by further studies. Since novel therapies directed against some of the molecules mentioned above have already been approved for the treatment of pulmonary hypertension, or, at least, are currently under investigation in clinical trials, we believe that caveolins might offer an interesting molecular target for novel therapeutic strategies as well. Along this line, HMG-CoA-reductase inhibitors (statins) were shown to alter the distribution pattern of the cholesterol-enriched caveolins [7] and their application has been shown to ameliorate pulmonary hypertension in several animal models [24, 25].
The “classical” morphological features of pulmonary hypertension such as the formation of a neo-intima and marked hyperproliferation of the smooth muscle cell layer have not been observed in the present study. This might be due to the fact that the increase in pulmonary vascular resistance in COPD-related pulmonary hypertension is generally very moderate. Our study is further limited by the lack of a control group consisting of patients without COPD and the relatively small number of patients. Nevertheless, the findings presented herein are in line with the data from Patel et al, and therefore strongly suggest a common molecular pathway involving caveolar proteins in the development of pulmonary hypertension both in its idiopathic form and in relation with advanced COPD.
In summary, we show for the first time that the expression pattern of Cav-1 within pulmonary arteries is associated with pulmonary hypertension in COPD patients. Consistent with the results from studies performed in tissue samples derived from idiopathic pulmonary arterial hypertension, the expression of Cav-1 was found to be correlated with the pulmonary arterial pressure. Since caveolin has been associated with different molecular players in pulmonary hypertension and, moreover, appears to regulate fibrosis a least in part, our results emphasize a potential novel factor in the pathogenesis of COPD-associated pulmonary hypertension.
ACKNOWLEDGEMENT
We thank the staff members of the Institute for Surgical Pathology, in particular M. Baucamp and S. Behnke for excellent technical assistance in manufacturing serial sections and immunohistochemical stainings. All authors have been supported by their respective institutions.

