All published articles of this journal are available on ScienceDirect.

Proline-Glycine-Proline (PGP) and High Mobility Group Box Protein-1 (HMGB1): Potential Mediators of Cystic Fibrosis Airway Inflammation
Authors Info & Affiliations
Abstract
Cystic fibrosis (CF) is chronic lung disease characterized by an unrelenting neutrophil-predominant airway inflammatory response. This inflammation leads to extracellular matrix (ECM) remodeling and eventually to the development of bronchiectasis. While many components of the immune response in CF have been well-characterized, recent data suggests that small molecules may play an important and underappreciated role in this inflammation. This review will examine two novel molecules: proline-glycine-proline (PGP) and high mobility group box protein-1 (HMGB1), and their potential impact in CF lung disease. This review will provide a brief overview of CF lung disease and background on both HMGB1 and PGP. It will then focus on these molecules in a murine model of CF-like airway disease and in human biological specimens from CF individuals. Finally, this manuscript will address possible mechanisms for therapeutic targeting of these bioactive mediators.
INTRODUCTION
Cystic fibrosis (CF) is a common lethal genetic disease in persons of northern European descent, with a carrier frequency in this population of 1:25 and an incidence of ~1:3,000 births. Over 30,000 CF-affected individuals have been identified in the U.S., and the worldwide prevalence is estimated to be ~70,000 [1]. CF is caused by defective function of the CFTR gene product named the Cystic Fibrosis Transmembrane conductance Regulator, or CFTR. The disease is characterized by abnormal transport of sodium (Na+) and chloride (Cl-) across the epithelia of many tissues, including the lungs [2]. This dysregulated ion transport leads to a significant reduction of the airway surface liquid (ASL) of epithelial cells, leading to worsening mucus plugging and airway inflammation. Over time, this pathophysiology leads to ongoing airway remodeling with the development of progressive bronchiectasis and worsening lung ventilation/ perfusion mismatch [3]. These pulmonary manifestations of CF are responsible for substantial morbidity and disease-related mortality, with a median survival of approximately 37 years [4].
A notable result of the loss of CFTR function is the development of chronic airway inflammation. The airways of CF patients, while normal at birth, quickly become inflamed and chronically colonized with characteristic bacterial pathogens [5]. Inherent inflammation or early bacterial colonization may initiate this process, with the activation of TLR receptors on airway epithelia and neutrophils (PMNs) [6, 7] and subsequent activation of NF-kappa B-mediated inflammatory response [8]. This, in turn, leads to the release of prominent levels of IL-8 and IL-1 beta in the CF airways [9]. As delayed mucociliary clearance continues, persistent bacterial colonization leads to an aggressive and exaggerated inflammatory response with the ongoing influx of PMNs into the airways. PMNs release a variety of inflammatory products (such as myeloperoxidase, lactoferrin, and various proteases) causing airway epithelial damage and extracellular matrix remodeling [10-12]. This release can be carefully coordinated or may be as a result of PMN apoptosis or necrosis [13]. In addition to PMN influx, both tissue and alveolar macrophages are activated with airway bacterial colonization, leading to the release of a host of pro-inflammatory mediators [14].
Although many of these components of the innate immune response in CF lung disease have been characterized in numerous studies, other inflammatory pathways have been suggested as having potential importance. Recently, our group has highlighted two inflammatory molecules, originally described in other conditions, which may play an important role in CF lung disease. Both of these molecules (proline-glycine-proline (PGP) and high mobility group box-1 (HMGB1)) are found in an important animal model of CF-like lung disease and also in clinical samples from subjects with CF. Even though it is unknown as to how significant of a role these molecules may play in the overall inflammatory milieu observed in the CF airway, a recent publication by our group suggests that PGP is at least as important as IL-8 as a neutrophil chemoattractant in patients undergoing bronchiolitis obliterans syndrome after lung transplantation [15]. Similarly, in sepsis HMGB1 seems to be a potent and important mediator in the latter periods of this condition.
PGP BACKGROUND
It has been known for over 30 years that digested collagen may have the capacity to augment both the inflammatory and injury response. Postlethwaite et al. initially described that collagen fragments derived from type I, II, and III collagen have the capacity to cause fibroblasts to migrate in vitro [16]. This group also demonstrated that type I collagen fragments were chemotactic for monocytes in vitro [17]. Senior and colleagues have also described in vitro neutrophil chemotaxis to sequences found in type IV collagen [18]. This work was extended when Riley et al. demonstrated an increased neutrophilic response in rats after intratracheal administration of collagen fragments [19]. However, very little work has been done in examining how specific motifs in collagen elicit particular inflammatory responses until the PGP story was delineated.
The story of PGP peptides and their mechanisms of eliciting inflammatory response had its origins in the eye. Alkali injury to the eye causes a profound influx of PMNs and it is these PMNs which drive the inflammatory response leading to corneal ulceration and perforation. In 1995, Pfister and colleagues demonstrated that alkali degradation of whole cornea generated two tri-peptides, N-acetyl-PGP (N-α-PGP) and N-methyl-PGP that are chemotactic for neutrophils [20]. Injection of these peptides into normal corneas recapitulated alkali eye injury [21]. Thus, specific chemotactic peptides for neutrophils were identified but their mechanism of action remained unknown.
Further studies from our group have identified N-α-PGP as a neutrophil chemoattractant in a murine model of intratracheal LPS administration. Sequence and structural analyses revealed a similarity between the glutamate-leucine-arginine (ELR) positive CXC neutrophil chemokines, collagen fragments, and N-α-PGP. Chemotaxis experiments verified that the tripeptide is active on human PMNs and acts through the same CXC receptors (CXCR) as is well characterized for known CXCR ligands. Most importantly, N-α-PGP was detected in bronchoalveolar (BAL) fluid of COPD patients which had significantly higher concentrations of the tripeptide compared to samples from healthy control patients. Although not all COPD patient BAL fluid was positive for N-α-PGP, the patients who had elevated concentrations had radiographic evidence of emphysema and lower FEV1 values [22]. In all, these studies linked N-α-PGP with neutrophil influx resulting from ECM degradation during airway inflammation.
In addition to data suggesting the presence of PGP in clinical COPD BAL samples, our group has also demonstrated the presence of N-α-PGP and/or the non-acetylated form of the peptide (PGP) in murine models of pneumonic tularemia [23] and corneal inflammation [24], and clinical disease samples from patients with bronchiolitis obliterans syndrome after lung transplantation [15] and both serum and sputum samples from patients with COPD [25]. To our knowledge, this was the first matrix-derived cellular chemoattractant described in clinical human specimens.
HMGB1 BACKGROUND
High-mobility group box 1 (HMGB1) is an evolutionarily ancient protein which serves predominately as a deoxyribonucleic acid (DNA)-binding protein. It is ubiquitously expressed in the nuclear compartment of eukaryotic cells functioning as a transcriptional regulator via interaction of its A-box and B-box subunits with DNA. In the late 1990’s, H Wang, K Tracey and others reported that extracellular HMGB1 is released as a late mediator of inflammation in sepsis. HMGB1 was identified following stimulation of cultured macrophages with tumor necrosis factor-alpha (TNF-α) and IL-1, and was verified as an active mediator of sepsis when blockade of the protein in mice by specific antibodies following LPS-mediated sepsis significantly reduced lethality [26]. These findings were the first to suggest that HMGB1 may serve as a soluble mediator in the modulation of inflammation. As an extracellular protein, HMGB1 has pleomorphic effects including activation of NF-kB, diffuse endothelial activation, hepatocellular injury, epithelial leak, and systemic activation of inflammatory cells [27]. HMGB1 activates inflammatory cells through interactions between receptor for advanced glycation end-products (RAGE) or toll-like receptor (TLR)-2 and -4. As suggested by its kinetics in shock and the inflammatory response seen following HMGB1 challenge, other receptors are also activated by extracellular levels of HMGB1 [28], including those on neutrophils [29, 30].
Although principally expressed intracellularly, the active extracellular form of HMGB1 is released by monocytes upon activation by TNF-α [26, 31]. This occurs through the coordinated release from stimulated cells following acetylation of nuclear HMGB1, leading to cytoplasmic translocation and export via secretory vesicles [32]. In addition, active HMGB1 is selectively released from necrotic cells, whereas cells undergoing apoptosis largely (but not entirely) inactivate HMGB1 during programmed cell death [33]. Once released, HMBG1 has been reported to have proinflammatory effects described above, and high levels have been associated with human or experimental models of sepsis [26], hemorrhagic shock [34], rheumatoid arthritis [35, 36], systemic lupus erythematosus SLE [37], and within the airways during ventilator induced lung injury [38]. Table 1 summarizes characteristics of both PGP and HMGB1.
General Features of PGP and HMGB1
| PGP | HMGB1 | |
|---|---|---|
| Size | 3 aminoacid peptide | 30 kDa protein |
| Original description | Neutrophil chemoattractant | Intracellular transcription factor |
| Biological activity | Acetylated forms augments CXC receptor activation | Extracellular form activates TLR and RAGE receptors |
| Clinical relevance | Sputum and serum samples of CF, COPD, and BOS | Elevated levels sepsis, hemorrhagic shock, rheumatoid arthritis, and CF |
Abbreviations: CXC: cysteine-X-cysteine chemokine; TLR: Toll-like receptor; RAGE: receptor for advanced glycation endproducts; COPD: chronic obstructive pulmonary disease; BOS: bronchiolitis obliterans syndrome; CF: cystic fibrosis.
EVALUATION OF PGP AND HMGB1 IN A CF ANIMAL MODEL
One of the challenges in developing a comprehensive understanding of the inflammatory milieu of the CF airway is lack of availability of an appropriate CF animal model that links CFTR dysfunction and inflammatory lung disease [1]. CFTR knockout mice, and mice expressing the murine equivalent of F508del CFTR, exhibit gastrointestinal morbidity and mortality reminiscent of human meconium ileus, however do not exhibit significant pulmonary disease despite the presence of the ion transport abnormality in the upper and lower airway. Hyperexpression of the Beta subunit of the epithelial sodium channel (ENaC) serves as an alternative murine model (termed Scnn1b+-transgenic) that closely resembles the pathologic findings found in the CF lung, including hyperabsorbtion of sodium chloride, depleted airway surface liquid volume, mucus obstruction and stasis, and airway inflammation [39]. Similar to CF lung disease in humans, inflammation is neutrophil predominant, and associated with elevated murine chemokines KC, MIP-2, and LIX in the lower airways [39-41].
Despite the absence of chronic bacterial colonization, the extent of inflammatory activation observed in the Scnn1b+ transgenic mouse suggest activation of innate immunity plays a strong role in the pathogenesis of pulmonary inflammation in this CF model. As such, the Scnn1b+ transgenic mouse serves as an important model for defining the role of both HMGB1 and PGP in the CF airway. Recapitulating observations seen in CF sputum, Scnn1b+ transgenic mice exhibit increased concentrations of HMGB1 in BAL fluid supernatants by complementary assays of Western blot and ELISA, at concentrations known to induce neutrophil chemotaxis, receptor mediated proinflammatory effects, and observed in the serum of human sepsis subjects (~50 ng/mL). In addition, HMGB1 also has been shown to inhibit neutrophil mediated phagocytosis of apoptotic cells, an effect recapitulated by Scnn1b+ transgenic mouse BAL fluid, and blocked by inhibition of HMBG1 with neutralizing antibody. Moreover, intratracheal injection of wt mice with recombinant HMGB1 results in neutrophilic influx within 24 hrs, and resultant production of PGP, as also observed in the airway secretions of CF subjects (see also below) [40]. These findings suggest crosstalk between neutrophilic proinflammatory pathways governing HMGB1 and PGP in the CF airway, and raise the possibility that therapeutic intervention into either pathway could result in beneficial effects.
One interesting question that has evolved from the Scnn1b+ transgenic mice data is whether HMGB1 may serve to directly (or indirectly) generate PGP in vivo (Fig. 1). While it is unlikely that PGP is directly liberated from collagen by HMGB1, it is entirely possible that HMGB1 may induce the release of proteases from inflammatory cell populations, leading to the cleavage of PGP from intact collagen (Panel B). However, it is also possible that during necrosis of inflammatory cells, proteases are released along with HMGB1 and that HMGB1 has no direct influence on PGP production, but is rather a marker of cellular necrosis (Panel A). Clarification of these points may allow for an improved understanding if these pathways may overlap, highlighting potentially interesting therapeutic targets.
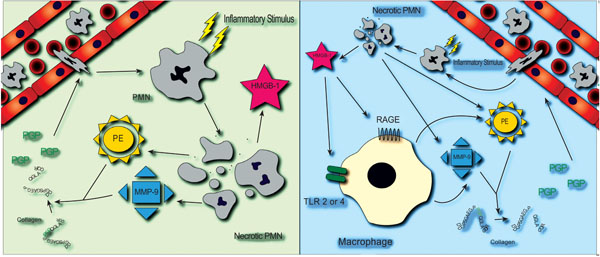
PGP and HMGB1 in the CF airway. Two potential pathways that may link airway expression of PGP and HMGB1 are shown. (A): Neutrophils are recruited to the airway by inflammatory stimuli and eventually undergo necrosis. Release of HMGB1 during necrosis is accompanied by concurrent but independent activation of proteases MMP-9 and prolyl endopeptidase, each required for the generation of PGP. (B): Extracellular HMGB1 activates airway macrophages through a TLR and/or RAGE dependent pathway, resulting in the release of MMP-9 and prolyl endopeptidase and generation of PGP, which perpetuate the inflammatory process.
ROLE OF PGP AND HMGB1 IN CF LUNG DISEASE
Due to the increased PMN burden and increased matrix destruction observed in CF lung disease, the potential presence and role of PGP in this condition became of significant interest. In CF individuals at the beginning of acute pulmonary exacerbation (APE), we found that 8 out of 10 individuals had both PGP and N-α-PGP in sputum samples compared to 1 out of 10 control, non-CF lung disease patients. In addition, through matched samples from individuals (n=16) at the beginning of and at the end of exacerbation, PGP levels declined during the course of inpatient hospitalization in 15 out of 16 individuals. The mean PGP levels went from 146 ng/ml to 80.0 ng/ml, although at hospital discharge levels were still 5-fold higher than those observed for non-CF lung disease patients. These results demonstrate that (a) PGP peptides are differentially expressed in those individuals with CF compared to those without CF and (b) in those CF individuals undergoing APE, PGP levels respond to therapy. These results suggest that PGP may be used as a biomarker during APE in CF lung disease [42].
Since it was suspected that PGP was hydrolyzed and released from collagen, it become important to define the proteolytic pathways that are involved in the release of PGP into the extracellular environment. Previously, our group (and others) had shown that during APE in CF lung disease, there is a notable increase in proteolytic activity observed in airway secretions when compared to either individuals with stable CF lung disease or individuals without CF [43]. However, to our knowledge, the only protease capable of releasing PGP from the collagen is prolyl endopeptidase (PE), a cytosolic serine protease predominately described in neuropeptide processing and angiotensin regulation [44]. While PE has been described in various pulmonary cell populations [45, 46], both its presence extracellularly and its role in pulmonary related pathology (including CF) were unknown.
Despite the identification of PE as a potential protease involved in PGP release, due to its β-propeller structure, PE could only act on substrates of 100 amino acids or less [47]. Since collagen is larger than this size restriction, liberation of PGP from collagen requires multiple proteases acting in concert. To test this hypothesis, mice were instilled intratracheally with various proteases known to be elevated in CF lung disease, either alone or in combination with PE, and then underwent bronchoalveolar lavage (BAL). The combination of MMP-9 and PE generated the largest quantity of PGP, with the combination of MMP-8 and PE also generating significant quantities. All other protease combinations did not generate PGP. The activities of all three of these proteases are elevated in CF sputum and there was a notable correlation between enzyme activity and PGP levels in clinical specimens. Due to this increased protease activity, when CF sputum is incubated on either type I or type II collagen, increased PGP levels are generated above those seen at baseline in CF sputum. However, when samples are pre-incubated with specific inhibitors of MMP-8, MMP-9, PE or combinations of these inhibitors, PGP generation is inhibited by 80-100%. These results not only highlight the generating system of PGP in vitro and ex vivo but also suggest the potential of targeting the generating proteases as a means of abating the effects of PGP in CF lung disease [42].
The intense neutrophilic inflammatory milieu found in CF, including the prominence of necrotic cells within the airway, strongly suggested that HMGB1 could also play an important role in CF airway disease, parallel to the findings in the Scnn1b+ transgenic mouse [48]. Indeed, levels of HMGB1 are elevated at baseline in CF sputum, and increase further during acute pulmonary exacerbation. Moreover, HMGB1 concentrations decrease with antimicrobial therapy directed at CF pathogens. Whether HMGB1 serves primarily as a marker of neutrophilic inflammation, or plays a direct pathogenic role has not yet been conclusively determined; however, potent effects of HMGB1, including induction of neutrophil chemotaxis and disruption of phagocytosis in vitro, and induction of lung injury in vivo, suggest it is at least a partial contributor to the disease [30, 40].
THERAPEUTIC IMPLICATIONS
Blockade of neutrophilic inflammation through innate pathways early in the pathogenesis of CF lung disease, prior to the onset of chronic bacterial colonization, may represent a strategy that obviates the potential risk of increasing infectious burden, as reported for a leukotriene B4 antagonist and methotrexate in CF individuals with moderate to advanced lung disease [49].
The kinetics of HGMB1 as a late mediator in sepsis, and its identified pathogenic role in rheumatoid arthritis, hemorrhagic shock, and acute respiratory distress syndrome, make antagonism of the protein an attractive potential therapeutic target [50]. Several potential strategies are currently under study. Proof of concept studies in experimental models have been shown in sepsis (including later forms of the syndrome [51]) and hemorrhagic shock [52], whilst rheumatoid arthritis can be mitigated with antagonism of HMGB1. The corticosteroid methylprednisonlone, known to have beneficial effects in CF lung disease [53], has been shown to antagonize HMGB1 [54], but is unlikely to be a viable option due to lack of specificity and excess toxicity. More specific inhibitors include anti-HMGB1 neutralizing antibodies, and recombinant A-box peptide, an HMGB1 domain that antagonizes the proinflammatory effects of the HMBG1 B-box domain through competitive inhibition [52, 55, 56]. In addition, inhibitors include cholinergic agents, ethyl pyruvate, nicotine, stearoyl lysophosphatidylcholine, and a number of Chinese herbs [55, 57, 58]. Recently, the flavinoid quercetin, a molecule class well known to potentiate the chloride channel function of CFTR, reduced circulating levels of HMGB1 in experimental mice with endotoxemia, an effect recapitulated in macrophage cultures [59]. Similarly, minocycline, a semisynthetic tetracycline, decreased HMGB1 expression in microglia cells, and was associated with a cerebroprotective effect in a murine model of ischemic injury [60].
Targeting PGP in vivo presents numerous opportunities and challenges. One potential mechanism to affect PGP levels in vivo is to quell the increased protease burden seen in CF lung disease. Our previous ex vivo data suggest that this is a very potent mechanism by which to modulate PGP; despite these results, most synthetic or semisynthetic specific MMP inhibitors utilized in clinical trials have either suffered from inadequate delivery to affected tissues or significant side effect profiles limiting their use [61, 62]. However, our ex vivo data also highlighted a significant decrease in PGP generation by doxycyline, a tetracycline antibiotic [43]. Thus the tetracycline class of antimicrobials represents a potential means to inhibit both PGP and HMGB1 pathways within the CF lung, and deserves further exploration. As such, there is an ongoing clinical trial in hospitalized CF patients to examining the effects of doxycycline as an anti-inflammatory therapy in CF lung disease which hopes to address some of these questions.
Another potential way to target PGP is by binding the peptide by either an antibody or small molecule. While PGP-specific depleting antibodies have been developed, the difficulties with generation of quantities of antibody for successful immunization and potential host hyperimmune responsiveness limit their potential usefulness as therapeutics. A small peptide, arginine-threonine-arginine (RTR) has been developed by our laboratory to physically bind PGP in vivo [63]. Recently, RTR was shown to inhibit both PGP and chronic LPS-induced pulmonary emphysema and right ventricular hypertrophy, suggesting that this peptide may be used in treatment of PGP-mediated inflammation in clinical disease [64]. Our laboratory has recently tested RTR with sputum from CF patients and has shown a notable decline in its neutrophil chemotaxis activity (Gaggar and Blalock, unpublished observations).
CONCLUSIONS
CF is an inflammatory lung disease characterized by intense neutrophilic inflammatory response. While many facets of this immune cascade have been studied, there is ongoing interest in delineation of novel effector molecules in this condition. The identification of PGP and HMGB1 offers a window into further enhancing our understanding of CF-related airway inflammation and highlights potential unique therapeutic opportunities in this condition. Further studies are needed to clarify the relative contributions that these molecules contribute to CF pathogenesis.
CONFLICT OF INTERESTS
The authors report no conflict of interest. This work was funded by the Cystic Fibrosis Foundation (GAGGAR07A to AG) and the National Institutes of Health (K23 DK075788 and 1R03 DK084110-01 to SMR) and (HL090999, HL077783, and HL087924 to JEB).
The content of is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute, National Institute of Diabetes, Digestive and Kidney Disease, or the National Institutes of Health.

