All published articles of this journal are available on ScienceDirect.

Asthma and COPD - The C/EBP Connection
Abstract
Asthma and chronic obstructive pulmonary disease (COPD) are the two most prominent chronic inflammatory lung diseases with increasing prevalence. Both diseases are associated with mild or severe remodeling of the airways. In this review, we postulate that the pathologies of asthma and COPD may result from inadequate responses and/or a deregulated balance of a group of cell differentiation regulating factors, the CCAAT/Enhancer Binding Proteins (C/EBPs). In addition, we will argue that the exposure to environmental factors, such as house dust mite and cigarette smoke, changes the response of C/EBPs and are different in diseased cells. These novel insights may lead to a better understanding of the etiology of the diseases and may provide new aspects for therapies.
1. ASTHMA
General background:
The term asthma origins form the greek word ‘asthmaino’ (αστημαινω), translated as ‘gasping’ and which was first used by Hippocrates (460-377 BC) in the Corpus Hippocraticum [1]. The Global Initiative for Asthma (GINA) defined asthma as a “chronic inflammatory disorder of the airways in which many cells and cellular elements play a role. The chronic inflammation is associated with airway hyper-responsiveness that leads to recurrent episodes of wheezing, breathlessness, chest tightness and coughing, particularly at night or in the early morning. These episodes are usually associated with widespread, but variable airflow obstruction within the lung that is often reversible either spontaneously or by treatment” [2]. This unifying definition of asthma highlights the clinical hallmarks of the disease: (i) the inflammatory process, (ii) the airway hyper-responsiveness, (iii) the obstruction of the airflow, and (iv) increased airway remodeling. Asthma is a very heterogeneous disease, as it includes immunopathology, clinical different phenotypes, non-uniform response to therapies and distinct natural histories [3]. Asthma can be considered as a syndrome with different risk factors, different prognosis, and different response to treatment [4, 5]. This indicates the need to rethink the definition of asthma as an inflammatory disease of the lung.
Although innate factors may have a genetic background, data analyses for genes associated with (1) increased production of IgE (atopy), (2) airway hyperrresponsiveness, or (3) the release of inflammatory mediators are rather inconsistent and provided no specific asthma-associated genes. However, a number of chromosomal regions were associated with asthma susceptibility and with the co-inheritance of the tendency to produce elevated IgE serum levels together with airway hyper-responsiveness [6-9]. In a genome-wide study, 79 genes were differentially expressed in cells of asthma patients relative to controls [10]. The expression pattern of these genes, however, was not straightforward but indicated complex interactions with several environmental risk factors. Known risk factors are obesity and male sex for childhood asthma [11]. Environmental risk factors that foster the development of asthma are indoor and outdoor allergens, such as house dust mite (HDM), cockroach allergens, cat and dog dander or Aspergillus mold. Especially, the exposure to allergens during childhood up to 3 years of age seems to be crucial for developing asthma-like symptoms [12-21]. Other risk factors are airway infections during childhood [22-25], occupational sensitisers [26-29], exposure to tobacco smoke [30-35], and the diet [36, 37]. There is considerable overlap of the mechanisms by which these risk factors may lead to the development of asthma. It is important to note that triggers for an asthmatic attack are not the same as factors that initiates asthma pathology. Many asthmatic subjects are atopic (60% of asthmatic adults, 80% of asthmatic children), but it is also true that not all atopic subjects develop asthma. Furthermore, 30% - 50% of asthmatic subjects are not atopic, meaning that no circulating IgE against one or more common allergens are present. Therefore, IgE-mediated mast cell degranulation is neither necessary, nor sufficient to develop asthma [38-40].
Pathogenesis of Asthma
Airway inflammation:
Airway inflammation is a multicellular process involving Th2 lymphocytes, eosinophils, activated mast cells, neutrohphils, macrophages and basophils. In atopic asthma, the airway responds to airborne inhaled allergens by a Th2 response with the release of a typical array of cytokines (Th2 paradigm) [3]. In the small and large airways of chronic asthma patients, the number of mast cells and airway smooth muscle (ASM) cells are increased. Activation of mast cells occurs after binding of IgE to the highaffinity IgE receptor (FCεRI) leading to the release of TNF-α, IL-4 and IL-5. Mast cell-derived bronchioconstrictive mediators, such as leukotriene (LT) D4, prostaglandin (PG)D2 and histamine, are potent ASM cell contractile agents [41, 42]. Vice versa, ASM cells produce chemokines, cytokines, and growth factors (e.g. IL-8, SCF, CXCL8, CXCL10) that further recruite mast cells into the lung [43, 44]. In addition, mast cell tryptase, a protease that activates protease activated-receptor type 2 (PAR2) on ASM cells induces muscle contraction [45].
Airway hyper-responsiveness:
Airway hyperresponsivness is a characteristic functional abnormality of asthmatic lungs resulting in airway narrowing when stimulated [2]. In consequence, asthma patients suffer from airflow limitation. Airway hyperresponsivenss is linked to both airway inflammation and and airway remodeling and is partially reversible by bronchodilators One of the mechanism leading to AHR may be an excessive contraction of the increased mass of ASM cells and by the thickening of the airway wall due to extracellular matrix.
Airway remodelling:
Airway remodeling in asthma includes thickening of the reticular basement membrane (RBM), epithelium fragility, hypertrophy of mucus secreting glands, hypertrophy and hyperplasia of ASM cells and increased deposition of extracellular matrix.
Damage and shedding of the airway epithelium is another important histological characteristic of asthma. Asthmatics patients often present clusters of epithelial cells (Creola bodies) in sputum and have increased numbers of epithelial cells in bronchoalveolar lavage fluid, which may result from the loss of airway epithelium observed in biopsy specimens [46, 47]. Upregulation of epidermal growth factor receptors (EGFRs), impaired proliferation reduced expression of proliferative markers and upregulation of the cyclin inhibitor, nuclear p21Waf1/Cip1 indicated that the epithelium is chronically injured and the repair mechanism is impaired in asthma [48, 49]. Furthermore, the epithelium of asthmatic patients is more fragile and the tight junctions are weakened [3, 50], in consequence, the airway epithelium enters into a chronic “wound scenario” [51].
Epithelial-derived growth factors force mesenchymal cells to produce collagen, reticular and elastic fibers, as well as proteoglycans and glycoproteins of the extracellular matrix (ECM), all of which contribute to the thickness of the airway wall in asthmatic [52]. The increased volume of the inner airway wall has functional consequences in terms of lumen reduction. Fibroblasts and myofibroblasts further contribute to tissue remodeling by deposing ECM components such as elastin, fibronectin, and laminin [53]. Mast-cell derived serine protease is a potent stimulant of fibroblast and ASM cell proliferation and stimulates the synthesis of type I collagen by human fibroblasts [54]. The distinct histologies of asthma are shown in Fig. (1).
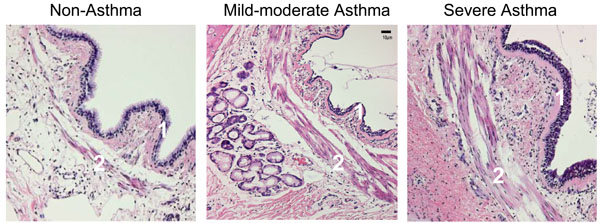
Immunohistochemistry of a representative section of the airway of a non-asthma control, a patient with mild-to-moderate asthma, and a patient with asthma who died of status asthmaticus (as indicated). The asthmatic airway demonstrates typical thickening of the basement membrane (1), and the characteristic increased mass of smooth muscle cells (2) [Reprinted with permission of the American Thoracic Society. Copyright (c) American Thoracic Society. Borger P, Tamm M, Black JL, Roth M. Asthma: is it due to an abnormal airway smooth muscle cell? Am J Respir Crit Care Med. 2006 Aug 15; 174 (4): 367-72; Official Journal of the American Thoracic Society, Diane Gern, Publisher].
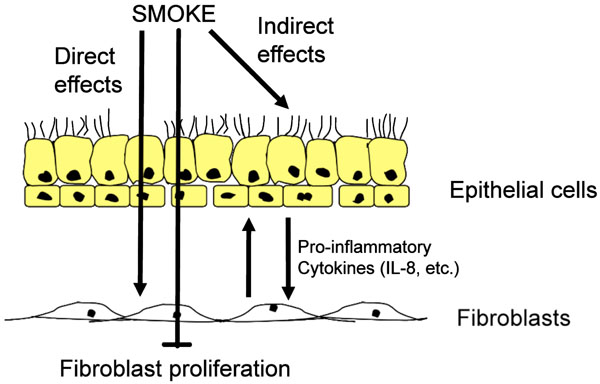
Effects of cigarette smoke. Cigarette smoke is able to penetrate the airway wall, thereby activating epithelial cells and fibroblast to produce proinflammatory cytokines, in particular interleukin 8 (IL8). In addition, cigarette smokes causes the fibroblasts to stop proliferation. Over time, the net effect may result in a loss of lung tissue.
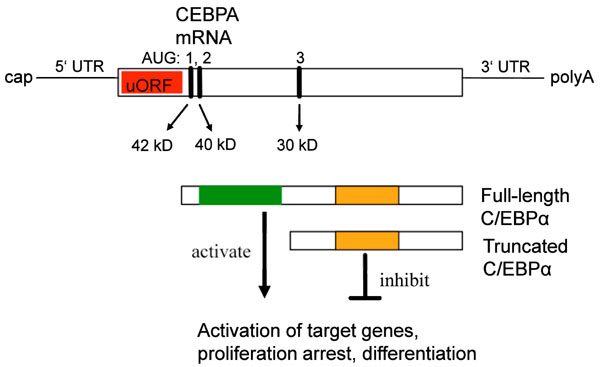
Schematic representation of the CEBPA mRNA, the position of the translation start codons (AUG), and the translation products (red: upstream open reading frame; green: transactivation domain; orange: DNA binding domain; uORF: upstream open reading frame).
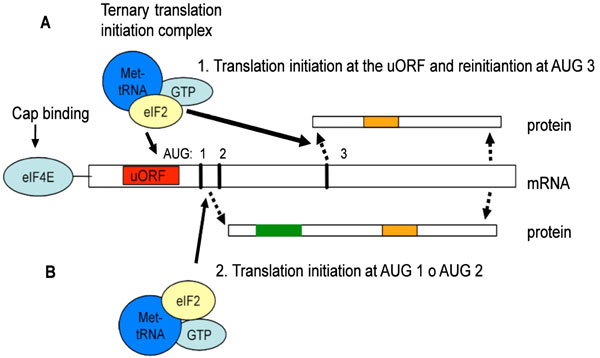
Schematic representation of the CEBPA mRNA translation mechanism, leading to the formation of the truncated or full-length C/EBPα protein. For truncated C/EBPα proteins, the translation starts at the uORF and stops after translation of the uORF. Then it reinitiates at AUG 3, generating the truncated isoform (A). For the generation of the full length C/EBPα the translation starts at the start codon AUG 1 or AUG 2 and reads trough (B). (Red: upstream open reading frame; Green: transactivation domain; Orange: DNA binding domain).
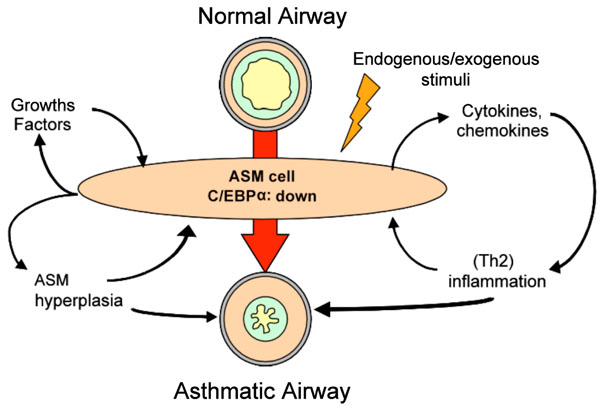
Model with the proposed central role of the airway smooth muscle (ASM) cell in airway inflammation and remodeling. Due to predisposition and/or environmental stimuli, ASM cells of patients with asthma express decreased levels of the C/EBPα. [Adapted from Borger P, Tamm M, Black JL, Roth M., Asthma: is it due to an abnormal airway smooth muscle cell? Am J Respir Crit Care Med. 2006 Aug 15; 174(4): 367-72].
The airway smooth muscle cell:
One of the most striking aspects of the pathology of airway remodeling in asthma is the increased number and size of ASM cells [55], which had been reported by Huber and Koesser in 1922. At that time, the ASM cells were considered the main cause of the airway hyper-responsiveness, and held responsible for the exaggerated airway constriction as observed in asthma [56]. Later, this concept was replaced by the hypothesis that asthma results from a deregulated immune response, which however can only explain allergic asthma. Interestingly, an increasing number of recent studies points back to the pathologic ASM cell as a major cause of asthma. What properties of the ASM cell would support this idea? As ASM cells are the effector cells controlling the airway caliber, it is reasonable to consider that dysfunction of ASM cells contributes to the pathophysiology of asthma. Today, asthma is defined as chronic inflammatory disease of the lung with an increased Th2-like response and with high levels of IL-4, IL-5, and IL-13 [2]. This view ignores that the increased mass of ASM cells already exists in very young children and does not correlate with the severity and duration of the disease [57-62]. Furthermore, airway inflammation is not present in all patients with childhood asthma, whereas remodelling is [59, 63].
As reviewed by Borger et al. [64], asthma is a heterogeneous disease involving not only immune-mediated mechanisms. Studies using cyclosporine to block T-cell activation and thereby the release of IL-2, IL-4 and IL-5 showed only an effect on the late asthmatic response, suggesting that the early asthmatic response is not T-cell mediated [65, 66]. The use of anti-IL-5 monoclonal antibodies reduced eosinophils by 80% in the airways and in the blood of asthmatic subjects but did not reduce in any clinical measures of asthma [67]. More recent studies, however, report beneficial effects of IL-5 antibody in eosinophilic asthma [68]. Interestingly the reduction of ASM cells mass by thermoplasty in the airway wall significantly improved asthma symptoms over a period of 3 years to date [69]. In the light of these findings, ASM cells may not be only effector cells that cause airway constriction as a consequence of the inflammatory process, but they may be initiator- or co-initiator of the disease. In line with this hypothesis, ASM cells produce IL-1, IL-2, IL-5, IL-6, IL-11 and IL-12, and are able to release TARC, a cytokine that induces Th2 migration and recruitment [70-74]. Therefore, ASM cell activity may initiate and orchestrate an inflammatory response and induce the recruitment of inflammatory cells into the lung. This capacity may be further enhanced through crosstalk with the airway epithelium.
Our group was the first to demonstrate that isolated ASM cells of asthmatic patients have the potential to proliferate faster than cells from control subjects under defined conditions [75]. This could explain the augmentation of ASM cell mass in the asthmatic airway. Importantly, this ASM cell pathology is maintained through weeks in culture and many passages long after any inflammatory mediator present in the tissue has been washed out. Furthermore, our group found that ASM cells from asthma patients are primed for IL-6 release [76, 77]. We can therefore speak of a constitutive activated phenotype of ASM cells in asthma patients.
2. COPD
General background:
COPD is a chronic inflammatory lung disease that will be the third most frequent cause of death throughout the world. The Global Initiative for Chronic Obstructive Lung Disease defines COPD as “a pulmonary disease characterised by airflow limitation that is not fully reversible. The airflow limitation is usually progressive and associated with an abnormal inflammatory response of the lung to noxious particles or gases” [78]. COPD affects >10% of the world population over the age of 40 years [79] and every year almost 3 million people die of this disease [80, 81]. Despite of its global prevalence, there is still a fundamental lack of knowledge about the cellular, molecular and genetic causes of COPD and an efficient therapy is non-existing [82].
COPD involves host-dependent (genetic) and environmental factors, but undoubtedly the major cause, is cigarette smoking; it accounts for approximately 90% of all cases and there is a correlation between the tobacco consumption and severity of COPD. Furthermore, indoor air pollution from biomass fuel smoke is a major cuse of COPD in the developing world [83]. However, only 10-20% of people exposed to smoke develop COPD, which indicates that a genetic susceptibility has to be combined with environmental factors to induce COPD [84-86]. Mutations in the alpha-1 antitrypsin gene, which speciies a serine protease, lead to the development of emphysema, and decline in lung function due to digestion of the lung forming extracellular matrix and cell-cell interactions [87]. Mutations in genes of the detoxification proteins GSTM1 and GSTT1, also confer a risk for declining FEV1, especially in males, and this risk is further increased by smoking [88]. In addition, gene polymorphisms of the ADAM33 [89], DECORIN and TGFB1 genes confer the susceptibility to develop COPD [90]. Environmental COPD risk factors are occupational dust and chemical exposure [91-94], infections [95-98], the socioeconomic status [99]. Others regard COPD as an auto-immune disease with an aberrant response to antigens (e.g. elastin) released after smoking-induced tissue injuries [100].
Pathogenesis of COPD
Airway inflammation:
The immune cells in the COPD lung are disease-specific with predominantly neutrophils, alveolar macrophages, and CD8 positive T-cells. Compared to “healthy” smokers, COPD patients with emphysema show a 25-fold increase in the number of macrophages in the lung tissue and in the alveolar [95] and macrophage numbers in the airways correlated with the severity of COPD [101]. In COPD the inflammation occurs mainly in the peripheral airways (bronchioles), the lung parenchyma, and the pulmonary vessels. Distinct from asthma, the bronchioles of COPD lung are obstructed and present with fibrosis [102-107].
Airway remodelling:
In COPD, airway remodelling leading to airflow limitation is mainly observed in the small airways and lung parenchyma. In pulmonary emphysema, one of causes leading to defective alveolar regeneration is due to the malfunction of fibroblasts [108, 109]. The major characteristic of airflow limitation in COPD is the fact that it is only partly reversible and progressive. In COPD, the thickness of the airway wall of small airway increases and thus limits the airflow. This is due to a high collagen deposition and mucosa thickening [110-114]. The increased ECM accumulation is thought to be a consequence of the chronic inflammatory process. Increased levels of TGF-β are typical for COPD lungs and may cause the release of connective tissue growth factor (CTGF) which, in turn, stimulates collagen deposition in the airway wall [115-118]. Furthermore, inflammation destructs the alveolar wall leading to airway wall deformation and narrowing of the airway lumen which may ultimately lead to loss of lung tissue and emphysema [119].
Protease-antiprotease imbalance:
The major hypothesis to explain COPD-associated emphysema is an imbalance of proteases and their inhibitors. This idea might result from the fact that congenital emphysema is caused by the deficiency of alpha-1 antitrypsin, a protein that prevents the lung tissue from being digested by proteases. The lack of this protein accounts for 2% of COPD patients, with a substantial higher risk in smokers [87]. Unopposed, the activity of proteases induce lung damage similar to tissue structural changes seen in COPD-associated emphysema. In addition, matrix metalloproteinases (MMPs), may be involved in COPD since they induce morphological changes in the lung and increased concentrations of MMP-1, -2, -9, -12 were found in bronchoalveolar lavage samples of COPD patients [120, 121]. Since MMP-12 degrades elastin it is considered the leading proteinase responsible for pulmonary emphysema [122].
Cigarette smoke:
Cigarette smoke is a complex mixture that consists of over 4,000 chemical components, including 1015 highly reactive molecules in the gas phase alone [123-125]. In vitro, the biological effect of tobacco smoke can be studies using single compounds (e.g. nicotine), or cigarette smoke extracts (CSE), or smoke conditioned medium (SCM) [126] (Fig. 2). Cigarette smoke inhibits alveolar repair and hence contributes to tissue loss observed in emphysema [127]. CSE-stimulated lung fibroblasts release neutrophil and monocyte-attracting factors, including IL-8, granulocyte-macrophage colony-stimulating factor (GM-CSF), and monocyte chemotactic protein-1 (MCP-1) [128]. Chronic cigarette smoke exposure increased the number of neutrophils, lymphocytes, and macrophages in the lung [129, 130]. Significant retention of activated neutrophils in the lung was observed after smoking [131]. In a mouse model, cigarette smoke increased desmosine in the lung, which indicates elastin fiber breakdown [132]. Compared to non-smokers, exhaled breath condensate of healthy smokers contained increased levels of IL-1β, IL-6, IL-8, IL-10, and TNF-α [133]. In rodent models, acute and chronic cigarette smoke exposure increased levels of TNF-α, IL-1β, IL-8, and MCP-1, and increased mononuclear cells and neutrophils in the lung [134]. TNF-α may be the crucial factor for cigarette smoke-induced emphysema, since overexpression of TNF-α causes emphysema and alveolar inflammation [135]. Likewise, TNF-α receptor knockout mice were protected against emphysema [136]. Finally, cigarette upregulates MUC5AC expression, amplifying the expression of respiratory mucins and reduce the airflow [137].
3. CEBPs AND CHRONIC INFLAMMATORY LUNG DISEASES
C/EBPs:
C/EBPs comprise a family of six proteins, C/EBPα, β, δ, γ, ε and ζ, which are characterised by two transactivation domains, a basic DNA binding domain and a leucine zipper motif. The latter mediates dimerization between same (homo-dimers) or other C/EBPs (heterodimers) [138, 139]. C/EBPs are pleiotropic proteins involved in inflammation, cell differentiation and tissue remodelling. C/EBPs are involved in the fine-tuning of cell differentiation and metabolism. Their ability to control differentiation is achieved trough their interaction with other transcription factors, such as the peroxisome proliferator activated receptor-γ (PPAR-γ) in a cell type specific manner [140, 141]. Due to functional redundancy, C/EBP- null mice often fail to yield informative phenotypes [140]. The best studied C/EBP family members are C/EBPα and C/EBPβ.
C/EBPα:
C/EBPα is a DNA binding transcription factor that binds to the CCAAT box motif present in several gene promoters [142]. The highest level of C/EBPα mRNA are found in differentiated cells of the liver, adipose tissue, the intestine, the lung, and the adrenal gland, as well as in myeloid and placental cells [143]. C/EBPα plays a crucial role in cell growth arrest and cell differentiation, showing an expression pattern which is inversely related to proliferation [144]. As reviewed by Johnson et al. [145], initial evidence for the anti-proliferative function of C/EBPα came from the activation of a chimeric C/EBPα-estrogen-receptor (ER) by estrogen-arrested pre-adipocytes in the G0/G1 phase [146]. More recently C/EBPα has been implicated in the development of a form of acute myelogenous leukemia (AML1-ETO), where C/EBPα expression is suppressed or strongly decreased, leading to leukemogenesis and impaired neutrophil differentiation due to an inhibition of cell cycle exit [147]. In acute myeloid leukemia (AML) the oncogenic fusion proteins BCR-ABL and AML1-MDS1-EVI1 increased calreticulin (CRT) levels, which then inhibited C/EBPα translation [148]. In general, the down-regulation of C/EBPα protein appears crucial in myeloid leukemia development. In the lung, C/EBPα is also a master regulator of airway epithelial differentiation and its loss of function as a tumor suppressor leads to non-small lung cancer [149]. C/EBPα associates with the cyclin-dependent kinase (CDK) inhibitor p21 [150], which then binds and inhibits the activity of CDKs. C/EBPα binds directly to CDK2 and CDK4 thereby inhibiting them to phosphorylate their substrate [145]. In normal cells, C/EBPα forms a complex with the glucocorticoid receptor (GR), which then activates the p21(Waf1/Cip1) promoter [151-153]. Furthermore, C/EBPα can directly repress S-phase driving genes either forming a complex with EF2 or binding directly to the CCAAT consensus site in these genes and suppress their transcription [145]. These findings show that C/EBPα protein plays an important role in both cell-cycle arrest and cell differentiation. However, C/EBPα can also have opposite functions. For instance, in p21-deficient mouse embryo fibroblast C/EBPα induced cell-cycle exit [154].
Translation control of CEBP mRNA:
In humans, the mRNAs of the CEBPA and CEBPB genes are present ubiquitously and protein levels are predominantly regulated at the translational level. The mRNAs of both genes have specific and highly distinguished regulatory motifs, and several C/EBPα proteins are translated from a single CEBPA mRNA by three different so called Kozak sequences [155]. The different C/EBPα isoforms retain different parts and functional domains and display opposite functions regarding gene regulation and cell proliferation. The full-length C/EBPα (p40/42) is a transcription, and contains a transactivation domain, while the truncated C/EBPα (p30) lacks this transactivation activity and counteracts the function of the full-length protein [156-159].
The mechanism of translation control of the CEBPA mRNA is presented in Fig. (3). CEBPA mRNA contains three translation initiation sites (AUG), which are in an optimal Kozak consensus sequence [155, 160]. The human CEBPA mRNA, therefore, can be translated into three proteins of different size: 42kD, 40kD and 30 kD proteins, respectively. Null-mutations abolishing the expression of the full-length forms [p42, p40] enhanced the expression of the truncated protein (p30) [155]. An additional upstream open reading frame (uORF), which is always out of frame with respect to the CEBP coding frame, translates a small pentapeptide. Mutations in this uORF abolished the translation of the truncated form (p30) and enhanced the expression of the full-length (p40/42), demonstrating that the uORF is essential for differential translation initiation. The organization of the CEBPB mRNA is very similar to that of CEBPA and the same regulatory mechanism generates full length and truncated proteins [155].
Several distinct pathways control the initiation of CEBP mRNA translation: (1) glycogen-synthase kinase 3 (GSK3), (2) phosphoinostitol 3-kinase (PI3K), and (3) mammalian target of rapamaycin (mTOR). The RNA-dependent protein kinase (PKR) is part of the GSK3-pathway and affects translation initiation by phosphorylation-induced inactivation of the rate-limiting translation protein eIF-2, which is part of the ternary eIF2/GTP/Met-tRNA-Met complex [161]. This facilitates the recognition of the AUG-codon and initiates protein synthesis. In addition to PKR, three more eIF2 kinases exist: (1) haem-regulated inhibitor kinase (HRI), (2) PKR-like endoplasmic-recticulum kinase (PERK) and (3) GCN2 [162]. In PKR mutant cells, eIF-2 cannot be inactivated and the C/EBPα expression shifted towards the truncated form. Similarly, activation of the mTOR pathway leads to more truncated proteins [155], as mTOR phosphorylates and inhibits PP2A, which in turn keeps the translation inhibitory 4E-BP1 protein in an active state [163-167]. It has been reported that 4E-BP1 inhibits eIF4E activity [168]. In addition, binding of eIF4E to the mRNA-cap is the rate-limiting step of the eIF4E complex and of the initiation of translation [169]. Over-expression of eIF4E shifted CEBPA mRNA translation towards the truncated isoform, while the inhibition of mTOR by rapamycin reduced the expression of the truncated C/EBPα isoform [155]. High eIF-2 and eIF-4E activity leads to predominant expression of truncated C/EBPα and C/EBPβ proteins.
The uORF is crucial to modulate the ratio of C/EBPα isoforms and thus the re-initiation of translation. When the translation activity from the uORF initiation codon is low, steric hindrance of the ribosomal complex is also low which generates full-length C/EBPα proteins, because translation is initiated from the first and second AUG-codon (see Fig. 4). Lack of nutrients or the presence of inhibitory signals, further decreases truncated proteins due to reduced efficiency of translation re-initiation at the third AUG codon [155]. When translation activity from the uORF initiation codon is high, however, steric hindrance is high and the third AUG-codon is the preferred site of translation initiation. This results in an increased level of truncated [p30] proteins [170, 171]. The efficiency of translation re-initiation depends on the reloading of the eIF2/GTP/Met-tRNAMet complex, whereas the eIF4E complex is required for efficient scanning and re-initiation after uORF translation [155, 172]. The rapid shift from truncated to full-length protein allows for a stringently controlled regulation and fine-tuning of cellular response to external stimuli. This is important, because it determines whether cells proliferate or exit the cell cycle and differentiate. The small uORFs is a cis-regulatory mRNA element of translation initiation, that has been found in various regulatory genes, including those for transcription factors SCL/Tal1 [173] and ATF-4 [174], thrombopoietin (TPO) [175], cyclin CLN3 [176] and beta-Secretase (BACE-1) [177].
Finally, CEBPA mRNA translation is reduced by calreticulin-binding to a CEBPA mRNA stem loop formed by a GC rich motif. Binding of calreticulin to this motif repressed the translation [178]. In adipogenesis, an inverse relationship of C/EBPα and calreticulin expression exists and calreticulin promoted adipogenesis by repressing the expression of C/EBPα and PPARα [179]. The opposite also occurs, when the hematopoietic zinc-finger, Hzf, interacts with the 3’ un-translated region of the CEBPA mRNA, thereby enhancing the translation [180].
CEBP mRNA translation and disease:
The etiologies of several human diseases can be traced back to mutations of genes that encode proteins of the translation control machinery. A wide range of proliferative disorders, including cancers, has been associated with deregulated or faulty mRNA translation [181]. Mutations of the PERK kinase that regulates eIF2 activity cause the Wolcott-Rallison Syndrome (WRS), a form of permanent diabetes [182-184]. Increased levels of eIF4E are found in several cancers [185, 186], including colon adenoma and carcinoma [187], breast carcinoma [188, 189], non-Hodgkin’s lymphoma [190] and primary bladder cancer [191], and in chronic myeloid leukaemia, the expression of the RNA-binding protein hnRNP is abnormally high. Interestingly, hnRNP binds to the 5’ UTR of the CEBPA mRNA thereby inhibiting its translation [192]. In addition, the deregulation of eIF2 is frequently observed in cancer cells [159, 187, 190, 193]. Congenital thrombocythaemia, a disease characterised by sustained proliferation of bone-marrow mega-karyocytes, is also caused by a mutation in the uORF of the cytokine thrombopoietin [174].
Airway remodelling and C/EBPs:
In human ASM cells and fibroblasts, C/EBPα regulates proliferation trough the induction of the cell cycle inhibitor p21Waf1/Cip1. In normal cells, β-mimetics and steroids activate p21Waf1/Cip1 via C/EBPα forming a complex with the glucocorticoid receptor (GR). In absence or at low levels, the C/EBPα complex with the GR can not be formed in sufficient amounts to activate the p21Waf1/Cip1 gene and may explain the increased proliferation of ASM cells. [151-153, 194, 195]. We recently showed that ASM cells of asthma patients have normal levels of CEBPA mRNA, but the translation is impaired leading to a decreased expression of the C/EBPα protein [196]. This mechanism operates via the uORF of the CEBPA mRNA [197]. In this respect, it is important to note that in a rhesus monkey model, exposure to HDM during the early years of life led to a persistent increase of ASM cells by an unknown mechanism but independent of the immune system [198]. Hence, hyperplasia of ASM cells may be the result of prolonged exposure to HDM allergens. Indeed, we recently observed that HDM extracts significantly reduced C/EBPα expression in ASM cells of asthma patients [197]. Interestingly, the reduction operated via the uORF mechanism, but rather via increased levels of calreticulin. In addition, HDM exposure induced ASM cell proliferation and enhanced the release of IL-6. Furthermore, HDM reduced C/EBPα levels via the activation of the PAR-2 receptor. Hence, HDM triggered both protease-dependent and protease-independent mechanisms that regulate C/EBPα [197]. These observations indicate a link between deregulated C/EBPα translation and in vitro hyperplasia of ASM cells and inflammation. The mechanism by which an impaired translation of the CEBPA mRNA may lead to the characteristics of an asthmatic airway is presented in Fig. (5).
Airway hyper-responsiveness and C/EBPs:
Airway biopsy specimens of asthma patients exhibited a significant increase in the contractile properties [199, 200]. Even single ASM cells demonstrated increased intrinsic contractile properties, which coincided with enhanced expression levels of myosin light-chain kinase messenger RNA in ASM cells of asthma patients [199]. Such an increase might account for the increased velocity of muscle cell shortening, since myosin light-chain kinase (MLCK) phosphorylates the regulatory light chain of myosin and regulates the rate of cross-bridge cycling, and therefore of the contractile properties of ASM cells. Although the promoter that regulates the expression of this kinase contains several C/EBP binding sites, the effect of C/EBP binding in this region is currently unknown [201].
Airway inflammation and C/EBPs:
NFκB is an important proinflammatory transcription factor required for the expression of cytokines, adhesion molecules, chemokines and growth factors [74, 202]. Interestingly, C/EBPα has the potential to silence this inflammatory response through interference with NFκB [34, 35]. The expression of many cytokines [203] and TARC [202] depends on NFκB and/or C/EBP binding sites in their promoters. Because C/EBPα (p40/42) is mainly a negative regulator of gene expression, a diminished expression of it may initiate airway inflammation through the release of proinflammatory mediators into the airway. The asthma-associated cytokine IL-4 blocks C/EBPα expression [204], hence sustaining airway inflammation. The observation that ASM cells of asthma patients produce less PGE2 than those from normals, suggests that PGE2 may also sustain a Th2-like inflammation. PGE2 is a potent inhibitor of proliferation and cell activity. Binding of PGE2 to its receptor generates cyclic adenosine mono phosphate (AMP), which counteracts the production of many proinflammatory cytokines and chemokines. Th2-like cytokines, in particular IL-4 and IL-5, are less susceptible to the inhibitory effect of cyclic AMP than Th1-like cytokines [205]. Intriguingly, the expression of the main regulator of PGE2 production (COX2) critically depends on C/EBPs. Here, we also do not know the effects of the various C/EBP isoforms [206].
COPD and C/EBPs:
The role of CEBP genes in the development of COPD is less clear. In lung epithelial cells of COPD patients, DNA-binding C/EBPβ levels are decreased relative to cells of ‘healthy smokers’ and might render the epithelium resistant to efficient regeneration [207]. In primary human lung fibroblasts, cigarette smoke regulated both C/EBPα and C/EBPβ via two distinct translational control pathways [197]. In the absence of serum, cigarette smoke induced full-length C/EBPα and -β proteins via the uORF mechanism, which coincided with decreased proliferation and increased expression of IL-8 (which is C/EBP controlled) [208]. In the presence of FCS, cigarette smoke decreased C/EBPα and -β via an induction of hnRNP E2. Here, the anti-proliferative role of full-length C/EBPα proteins may explain the reduced proliferation of fibroblasts, hence providing a basis to understand the destruction of the tissue and the increased airway remodelling observed in later stages of COPD. In addition, increased full-length C/EBPβ levels may result in reducing the elastin levels in alveoli, because C/EBPβ is a negative regulator of elastin transcription [209].
C/EBPs and Therapy
The treatment of the increased bulk of ASM cells as observed in the lungs of asthmatic patients may be through the modulation of the C/EBPα translation. This would require novel therapeutical strategies that target mainly the signaling pathways regulating the translation control at the uORF, e.g. the mTOR pathway. Alternatively, lithium chloride has been shown to prevent the degradation of C/EBPα protein and might be tested as a candidate drug to counteract airway wall remodelling in asthma [210]. Similar strategies might be envisioned in the treatment of the remodeling processes in the lung of COPD patients, but with the focus on C/EBPδ [196].
4. GENERAL CONCLUSIONS AND OUTLOOK
The pathologies of asthma and COPD are genetically predisposed, but the environment plays a crucial role for the initiation and progression of these diseases. Impaired translation of the cell cycle regulators of the C/EBP family may play a significant role in the pathogenesis of these lung diseases. Here we argued that asthma may stem from an impaired translation of CEBPA mRNA. A direct interaction of ASM cells of asthma patients with HDM may be a key event that chronically reduces C/EBPα protein levels. The lack of C/EBPα protein speeds up cell proliferation and stimulates the release of pro-inflammatory cytokines. The results are an increased bulk of ASM cells and airway inflammation, two key pathologies in asthma. Regarding the development of COPD, cigarette smoke-induced aberrant translational control of CEBPA and CEBPB mRNAs may be the key to understand and treat COPD pathology. Today, no effective therapy for COPD is available and strategies to prevent remodelling parameters are scant. Restoring the balanced expression of both C/EBPα and C/EBPβ by intervention at the level of translation control may be beneficial for both asthma and COPD patients.
ACKNOWLEDGEMENT OF FUNDING
Swiss National Science Foundation (SNSF 320030-124905 and SNSF 310030-133109).
CONFLICT OF INTEREST
None Declared.

