All published articles of this journal are available on ScienceDirect.

Ventilator-Induced Lung Injury (VILI) in Acute Respiratory Distress Syndrome (ARDS): Volutrauma and Molecular Effects
Authors Info & Affiliations
Abstract
Acute Respiratory Distress Syndrome (ARDS) is a clinical condition secondary to a variety of insults leading to a severe acute respiratory failure and high mortality in critically ill patients. Patients with ARDS generally require mechanical ventilation, which is another important factor that may increase the ALI (acute lung injury) by a series of pathophysiological mechanisms, whose common element is the initial volutrauma in the alveolar units, and forming part of an entity known clinically as ventilator-induced lung injury (VILI).
Injured lungs can be partially protected by optimal settings and ventilation modes, using low tidal volume (VT) values and high positive-end expiratory pressure (PEEP). The benefits in ARDS outcomes caused by these interventions have been confirmed by several prospective randomized controlled trials (RCTs) and are attributed to reduction in volutrauma.
The purpose of this article is to present an approach to VILI pathophysiology focused on the effects of volutrauma that lead to lung injury and the ‘mechanotransduction’ mechanism. A more complete understanding about the molecular effects that physical forces could have, is essential for a better assessment of existing strategies as well as the development of new therapeutic strategies to reduce the damage resulting from VILI, and thereby contribute to reducing mortality in ARDS.
1. INTRODUCTION
The acute respiratory distress syndrome (ARDS) is a relatively frequent complication in critically ill patients and responsible for significant co-morbidity and mortality [1]. For many years, multiple studies have aimed to determine the mechanisms of ARDS generation and progression as well as other aspects including the recognition of subtypes of ARDS, identification of risk and prognostic factors and others, all areas that have intended to contribute to the development of new preventive and therapeutic strategies. Most of the studies show strategies to prevent the occurrence or progression of ARDS, and only a few interventions have focused on to reverse the damage or accelerate the recovery period caused by the primary injury. However and unfortunately, only a few studies have shown significant effectiveness that contribute to better clinical outcomes [1], specifically those whose targets were based on to reduce the ARDS progression through the development of protective strategies that result in a reduction of volutrauma.
The purpose of this review is to present an approach to the VILI pathophysiology mainly focused on the initial importance of the volutrauma effects in a previously healthy lung of patients afflicted with a "first hit" with risk of developing ARDS, as well as the effects of volutrauma in a lung with ARDS. A more complete understanding of these mechanisms may allow for the development of new therapeutic strategies to prevent VILI and consequently achieve better clinical outcomes.
1.1. Ventilator-Induced Lung Injury
Mechanical ventilation (MV) by itself has been established as a factor that induces lung damage, which has been demonstrated even in previously healthy lungs, where the MV could aggravate the lung effects of a ‘first inflammatory hit’, a concept known as ventilator-induced lung injury (VILI) [2-4]. VILI caused total lung damage, which is the result of the sum of local lung damage with a heterogeneous distribution due to heterogeneous local lung susceptibility. These local events promote an anisotropic alveolar inflation in the opening phase and collapse at the end of the expiratory phase, which promotes greater damage. This initial anisotropic distribution promotes a local inflammatory response in areas that receive the higher volutrauma, but subsequently these areas trigger a generalized inflammatory response in the lung tissue.
The pathophysiological basis of VILI are complex and characterized by different overlapping interactions [4], initially after the ARDS Network clinical trial [5], VILI was primarily linked to two phenomena: the first phenomenon occurs at the end of inspiratory cycle and is associated with the use of a high inspiratory pressure of tidal volume (VT) that promotes an alveolar over-distension, a process related to the risk of rupture of the airway and alveolar walls in the non-dependent lung regions (R-ND). The second phenomenon unfolds in dependent lung regions (RD), which are exposed to significant stress when the airway and alveoli open on inspiration and collapse on expiration tidal recruitment. This phenomenon is associated with the use of insufficient PEEP to prevent alveolar collapse and cyclical reopening. The two phenomena may overlap, since an unnecessarily high PEEP may lead to over-distension and an excessive VT drop, it can increase the effects caused by the cyclic alveolar collapse and reopening [5]. Moreover, some experimental studies have confirmed that the collapse of the airspace and the cyclical recruitment in lungs affected by a previous inflammatory conditions are crucial in the development of VILI [4, 6].
2. Mechanism associated with induction of volutrauma
2.1. Physical Forces
The volutrauma effects over the lung parenchyma can be evaluated at different spatial scales, in this sense the response of its resident cells to the deforming stress take importance in order to provide the biological basis of lung protective care.
2.1.1. Regional Mechanics in Lung
Physical key events associated to volutrauma are alveolar collapse and lung distension [7]. Alveolar collapse is determined by cyclic recruitment and derecruitment (’opening’ and ‘closure’) that could lead to damage of the epithelial lining by interfacial forces [8]. Indeed, in injured lung regions, as end-expiratory lung volume is quite low in the recumbent posture, the alveoli and conduction airways are particularly susceptible to closure induced by liquid plugs [9].
A protective approach that limits the lung damage must maintain sufficient end-expiratory volume to prevent alveolar collapse or open a collapsed/closed lung region [7]. To open a collapsed/closed lung region required a higher pressure than the pressure at which it was closed [10], then subsequent breaths preferentially inflate the remaining open units, so that gradually a ‘shrinking baby lung’ could accommodate the ventilatory demand [11]. The lung injury effects caused by the collapse and closing of alveolar could be reduced when lung areas may be permanently open [12, 13].
Mechanisms of biophysical injury associated with alveolar overdistension are responsible for lung injury through several effects including a decline in the surface tension due to reduction in surfactant properties [14], an increase of filtration coefficient and protein permeability in the endothelial and epithelial barriers [15], as well as wounding of alveolar resident cells by tensile and interfacial stress [8, 16]. In conclusion, a protective approach, must to reach an end-expiratory volume with a balance between preventing alveolar collapse and limit lung distension.
2.1.2. Mechanics of a Single Alveolus
At the alveolar level, the tidal stretches inflate the alveolus, therefore cells that lining the alveolus and their elastic linkages (the e-cadherins), become stretched. From physical perspective, the epithelial cells and their linkages through e-cadherin could be considered as springs in series. The capacity of the individual extension of these components depends on their relative stiffness, therefore if cells are too stiff, the intercellular linkages will drive the deformation during the inflation of the alveolus [7]. The stiffness of the epithelial cells is mostly determined by the actin cytoskeleton [17], whereas the total stiffness of e-cadherin linkages depends on their density around the cells being in parallel with each other’s [7]. Therefore, any pathology that affects an adequate metabolic functioning of epithelial cells could result in inadequate distribution of the cytoskeleton, which in turn lead to increased cellular stiffness, determine a higher stress on the intercellular junctions and alteration of the alveolar permeability.
2.2. Lung Stress and Strain
Unfortunately the values of lung deformation (or strain) that can be safely reached during mechanical ventilation are poorly defined, especially in humans [18]. In the clinical practice usually the lung distension is commonly inferred from VT (mL/kg of ideal body weight) and end-inspiratory (plateau) airway pressure [5]. However, neither of these two variables reliably reflects tissue deformation, especially during ALI (acute lung injured)[18], when relationships between body weight and functional residual capacity (FRC) and between airway and transpulmonary pressure become unpredictable [19]. From the physical view, strain and stress are used to describe the responses of a body to a external overload; strain is the relative change in size and shape and stress is the internal tension [20]. The exact measurement of lung strain and stress is infeasible in clinical practice, but it is possible make some approximate measurements of these.
Lung strain is the ratio between the volume of gas inflated (change in volume) and FRC [18]. On the other hand, lung stress equals the corresponding change in transpulmonary (airway minus pleural) pressure [21]. However, as the lung stress–strain relationship is almost linear until reach the total capacity [19], lung strain can be derived from stress and viceversa. In this fact, the lung strain can be computed from FRC, which several ventilators automatically measure it, whereas over-imposed lung stress can be estimated from changes in the esophageal pressure [19]. In the assessed of the strain effects, we must consider different types of strain: static, dynamic and global strain. For example, during mechanical ventilation without PEEP, lung deformation is due to tidal ventilation, so this dynamic strain can be computed as VT/FRC. Moreover, when PEEP (and the corresponding volume of gas, VPEEP) is applied, lungs are also kept tonically inflated above their FRC and are exposed to an additional static strain (VPEEP/FRC). Finally the global strain is the sum of these two components: global strain = (VT +VPEEP)/FRC.
2.2.1. Dynamic Versus Static Strain: Animal Studies
Most of the studies have shown that ventilation with large VT and zero end-expiratory pressure is more injurious than ventilation with small tidal volume and some PEEP, for a given peak pressure [22]. Some studies have shown that medium-sized animals ventilated with dynamic strain of 2.5 (static strain equal to 0), cause lung damage and develop fatal pulmonary edema. By contrast, those animals ventilated with smallest dynamic (0.5) and largest static (2.0) strain ended the experiment with normal lungs, although they needed higher amount of fluids and high dose of norepinephrine to maintain an adequate cardiac output [23]. In these medium-sized animals, harms of mechanical ventilation depend not only on overall lung inflation, but also depend of the form that this is achieved: large cyclic deformations are more injurious than equivalent, mainly static.
On the other hand, other studies in small animals showed apparently contradictory results [24], because healthy rats developed pulmonary edema when they were ventilated with small VT(7 ml/kg of body weight) and high PEEP (15 cm H2O). However, a methodological point of this latest study to consider was that lungs were inflated above their upper physiological limit [25]. These different results can be explained, being that the end-inspiratory volume does not exceed the total capacity, healthy lungs better tolerate static than dynamic, inflations [18]. However, when end-inspiratory lung volume exceeds the total capacity, static or dynamic inflation invariably causes lung damage. This findings support the measuring of lung volumes during mechanical ventilation [26].
2.2.2. Dynamic Versus Static Strain: Human Studies
The major survival benefits evidence by clinical research has been represented by limiting the VT and by the increasing PEEP during acute respiratory insufficiency [18]. In patients with acute respiratory failure, the limitation of VT is beneficial either per se [5] or in conjunction with increasing PEEP [27, 28]. By contrast, the only increase in PEEP generally does not diminish hospital mortality [3, 29, 30], unless that the VT is concomitantly reduced [27, 28]. Moreover, in patients with healthy lungs, although few clinical studies have been conducted, the conclusions are very similar [18].
First, one meta-analysis suggests that MV with smaller VT and higher PEEPs is associated with a lower incidence of pulmonary complications in patients without ALI at enrolment [31]. These findings have been confirmed for two subsequently RCTs [32, 33].
Second, in patients with non-pulmonary disease was also found that increasing the PEEP without changing the tidal volume did not diminish the incidence of ARDS in mechanically ventilated [34].
Lung damage during ventilation mainly depends on the amplitude and the velocity of dynamic tissue deformation according to suggested by experimental and clinical evidence and could be explained by tissue viscoelasticity. However, if heat produced by the internal frictions can change the conformation of molecules forming lung parenchyma, which is rich in water, is uncertain and needs to be validated in the future [18]. For the previously mentioned reasons, it has tried to develop strategies to minimize tidal ventilation, including ‘apnoea’ with extracorporeal carbon dioxide removal [35].
Animal studies [36] and the failure in high-frequency jet ventilation (at low PEEP)[37] suggest that a high static inflation (up to total lung capacity) may be required to maintain the lung open. This effect is due to that during apnea, bronchial secretions are not efficiently cleared, distal airways get obstructed and re-absorption atelectasis leads to progressive alveolar de-recruitment [18]. However, in two clinical trials in which tried to evaluate this idea using high-frequency oscillatory ventilation, these did not report benefit in outcome patients with ARDS [38, 39].
These findings could be explained by a right heart dysfunction because of higher mean airway pressure (close to or even above 30 cm H2O), with higher need for fluids and vasoactive drugs in the study population [40]. Therefore, it seems that interventions designed to protect the lung will hardly improve survival if they concomitantly harm the heart [18].
2.3. Influence of Ventilation Pattern
Beneficial effects have been found in animal models with an increased PEEP and a decreased VT by a reduction in the dynamic component of lung stretch [23]. These event is consistent with cellular studies suggesting that large stretch amplitudes and/or high strain rates are injurious to epithelial [41] and endothelial cells [42].
The risk of developing lung injury could be reduced with higher PEEP, which could open lung regions and make more homogeneous the stiffness at the length scales of small airways and alveoli [43]. In the intracellular level, structures could tolerate the static stretches of the PEEP by proper cytoskeletal remodeling, but are unable to adjust to the fast dynamically changing large strains [7]. Other factors that determine the effect of ventilation patterns in patients are high flow rates [44] and frequent deep inspirations [45], which can also worsen the ventilatory outcomes. In this regard, deep inspirations could help with recruitment and survival compared with conventional ventilation at 12 mL/kg [46], they could also have negative consequences [7]. The reason for these events could be since the integrity of the epithelium is compromised by deep inspirations resulting in increased e-cadherin levels in the lavage fluid [47]. Additionally, in lungs with edema, deep inspirations also caused cell apoptosis and alveolar capillary injury [48]. However, the mechanism that supports these findings are unclear, but they could also be related to the injurious effects of maximum strain and/or high strain rate on cells and intercellular junctions in the presence of high interfacial stresses [7].
In case of edema, deep inspirations also caused cell apoptosis and alveolar capillary injury [48]. The mechanisms behind these findings are unclear, but we speculate that they may also be related to the injurious effects of maximum strain and/or high strain rate on cells and intercellular junctions in the presence of high interfacial stresses.
Novel ventilation strategies have emerged trying to reduce the effects already described that lead to lung injury [49]. In the variable ventilation, VT changes are from cycle to cycle [49], and it has shown those not only improves lung function [12, 50-53], but also reduce the epithelial injury with a reduction of e-cadherin levels in lavage fluid [47]. These events could be explained by the ability of this ventilatory modes to maintain a better alveolar recruitment than conventional ventilation [54], and an important mechanism could be due to the variable stretching of epithelial cells that directly stimulates surfactant secretion [55], which should have a protective effect against edema formation but this need to be confirmed experimentally [7].
2.3.1. Ventilation at High Lung Volumes
High (absolute) lung volumes may cause lung damage by several mechanisms such as alveolar rupture, air leaks, and gross barotrauma (e.g., pneumothorax, pneumomediastinum, subcutaneous emphysema)[56]. The critical variable leading to the air leaks is regional lung overdistension, not high airway pressure per se, so the term of barotrauma can be misleading [57].
Lung overdistension may also cause more subtle forms of damage manifested by pulmonary edema [57]. The overdistension has been demonstrated in animal models, where ventilated rats with very high peak airway pressures (and therefore overdistention) and zero PEEP developed hypoxemia and postmortem examination revealed perivascular and alveolar edema [58]. Conversely, in rats that underwent ventilation with the same peak airway pressure but with the addition of a PEEP of 10 cm of water did not develop edema, which showing an interaction between overdistension and low end-expiratory lung volume with respect to lung injury [58]. However, the precise mechanisms underlying this interaction have not been completely elucidated [57].
Other animal studies have shown that the volume would have a major importance than airway pressure in the development of lung injury [57]. In this view, animals undergoing ventilation with high VT developed pulmonary edema while animals undergoing ventilation with similar airway pressures but with straps around their abdomens and chests that reduced the tidal volumes, did not developed pulmonary edema [59]. Experimental approaches showed that the volume (i.e., lung stretching), not
Airway pressure, was the most important factor in determining injury, a finding that led them to coin the term “volutrauma” [57].
2.3.2. Ventilation at Low Lung Volumes
Ventilation at low (absolute) lung volumes could also cause injury through multiple mechanisms, such as repetitive opening and closing of airways and lung units [60, 61], effects on surfactant function [62] and regional hypoxia. In this case, epithelial sloughing, hyaline membranes and pulmonary edema characterize lung injury caused, and it has been termed “atelectrauma” [60].
Moreover, the atelectrauma is amplified in lungs, in which there are marked homogeneities in ventilation [57]. Since many decades ago, it was shown that the stretching forces in lung parenchyma at margins between aerated and atelectatic regions could be up to four to five times as high as those in other lung regions [63].
3. MECHANISM OF VENTILATOR-INDUCED LUNG INJURY IN HEALTHY AND ARDS LUNGS
3.1. General Concept
The pathophysiological principles of VILI are complex and characterized by different overlapping interactions of multiple factors, some of which were previously described. These interactions include [4]: (i) high VT causing over distension; (ii) cyclic closing and opening of peripheral airways during tidal breath resulting in damage of both the bronchiolar epithelium and the parenchyma (lung strain), mainly at the alveolar-bronchiolar junctions; (iii) lung stress by increased transpulmonary pressure); (iv) low lung volume associated with recruitment and de-recruitment of unstable lung units (atelectrauma);(v) inactivation of surfactant by large alveolar surface area oscillations associated with surfactant aggregate conversion, which increases surface tension [64]; (vi) local and systemic release of lung-borne inflammatory mediators, namely biotrauma [65].
3.2. New Pathophysiological Concepts
Some experimental and clinical studies have demonstrated two main mechanisms leading to VILI: direct trauma to the cell, which promotes releasing of cytokines to the alveolar space as well as to the systemic circulation and also, the so-called ‘mechanotransduction’ mechanism.
3.2.1. Direct Volutrauma
The direct effect caused by volutrauma result in a persistent cyclic stretch during mechanical ventilation, which stimulates alveolar epithelial and vascular endothelial cells through mechano-sensitive membrane-associated protein and ion channels [66]. This cyclic recruitment of unstable overdistended alveolar regions provoked by MV causes initially a local inflammatory response, that is associated with the cellular necrosis surrounding lung tissue [66], and then a systemic inflammatory response which ultimately can lead to severe MODS (multiple organ dysfunction syndrome) [67].
The tissue deformation secondary to volutrauma activates nuclear factor-kappa B (NF-κB) signaling, which consequent leads to the production of interleukin (IL)-6, IL-8, IL-1β and tumor necrosis factor TNF-α [66]. For example, high IL-6 levels have been detected in various experimental models of VILI [68], and in the serum and bronchoalveolar lavage fluid of patients with ARDS under MV [67]. Thus, cytokines and chemokines are potential effector molecules that modulate and regulate VILI initial mechanisms. In this view, the effects of high VT ventilation even have been demonstrated in models without previous lung injury, where for example a high VT ventilation led to an increase in expression of intrapulmonary TNF-α and macrophage inflammatory protein-2 in mice [69].
3.2.2. Mechanotransduction
The term mechanotransduction is referred to the conversion of mechanical stimuli to a biochemical response when alveolar epithelium or vascular endothelium is stretched during mechanical ventilation [4]. Mechanical stimulus causes expansion of the plasma membrane and triggers cellular signaling thorugh various inflammatory mediators that influence a pulmonary and systemic cell dysfunction [66]. A high level of mechanical stretch is associated with increased epithelial cell necrosis, decreased apoptosis and increased of IL-8 levels [66]. Moreover, extracellular matrix (ECM), that determines the biomechanical behavior of the lung and plays a role in stabilizing lung matrix and fluid content, could also be affected [4]. This is because the mechanotransduction causes a mechanical force on ECM, which in turn causes a lung strain (the ratio between VT and functional residual capacity) [4]. Thus, high-VT ventilation causes ECM remodeling, influenced by the airway pressure gradient and the pleural pressure gradient [65, 70].
It has been in animal models that pulmonary edema formation in VILI develops when lung strain is greater than 1.5–2-fold [71]. Cyclic mechanical stress causes release and activation of matrix metalloproteinase (MMP), which play an important role in regulating ECM remodeling and VILI [4]. Moreover, lung strain could also lead to a modification of proteoglycan and glycosaminoglycans [4], which could cause different effects. For example, fragmentation of glycosaminoglycans may affect the development of the inflammatory response by interacting with various types of chemokine and acting as ligands for Toll-like receptors [70, 72].
Finally, mechanical strain induces ECM breakdown [70], which has demonstrated to be a signal for matrikines [4]. The relationship between pathophysiological events presented in VILI and mechanotransduction are shown in Fig. (1).
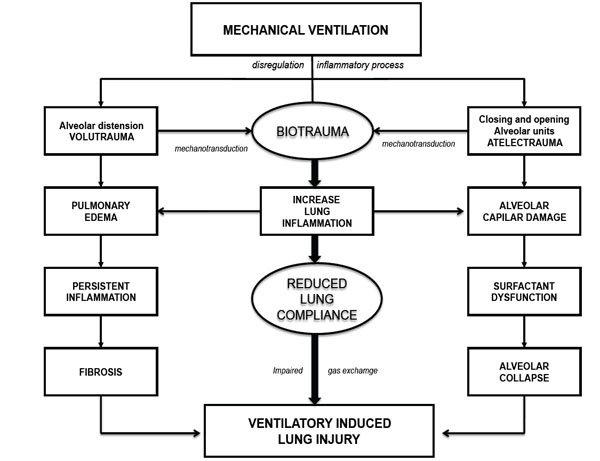
Mechanisms associated with VILI development.
4. VENTILATION STRATEGIES AND MOLECULAR EFFECTS
Multiple ventilation strategies have been evaluated with the purpose to reduce lung injury, for example: low tidal volumes to limit overdistention, higher PEEPs to prevent injury at low lung volumes (atelectrauma), and recruitment maneuvers (procedures used to reinflate collapsed lung units) through the sustained application of an airway pressure higher than approximately 35 cm of water, which if achieved inflating atelectatic regions, than minimizes the ventilation heterogeneity [57]. The clinical evidence of each of these ventilation strategies are briefly described below.
4.1. Low Tidal Volumes
Usually, ARDS patients have two types of regions: relatively normally aerated non-dependent lung regions and relatively non-aerated dependent lung regions, and this last case refers to regions that are lower from a gravitational perspective than other regions and therefore are more likely to be collapsed [57]. For this reason, the volume available for ventilation is smaller, so that this condition has received the term of "baby lung" [11]. Therefore a decreased VT, emerges as a solution to ventilate this "baby lung" and prevent overinflation of the relatively small, normally aerated regions [57].
This strategy was initially assessed by clinical studies [27, 73], and their results provided the basis for the further development of ARDS Network, the main study that evaluated the use of low tidal volumes [5]. The ARDS Network investigators compared a control strategy using a tidal volume of 12 mL/Kg of predicted body weight with respect to a low-tidal-volume strategy that used 6 mL/ kg of predicted body weight. The low-tidal-volume strategy was associated with an absolute reduction of rate of death [39.8% vs 31.0%] [5].
4.2. High PEEP and Recruitment Maneuvers
Several forms of respiratory failure are characterized by pulmonary edema and end-expiratory alveolar collapse, thus the prevention of a potential atelectrauma is essential for reducing lung injury. In this view, a low PEEP may be insufficient to stabilize alveoli and keep them open, thereby increasing the probabilityof VILI by atelectrauma [57]. A meta-analysis of randomized clinical trials in ARDS patients [74], concluded that a higher PEEP was associated with an absolute reduction of 5 percent in death rate among patients which had worse oxygenation, defined as a ratio of the partial pressure of arterial oxygen to the fraction of inspired oxygen [PaO2:FiO2] ≤ 200 mm Hg [74]. However, we should note that it is essential to find a value of PEEP not excessively high, because conversely, a too high PEEP could potentially cause adverse effects, including impairment of venous return and pulmonary overdistention [57]. For this reason, it is essential to develop a method to determine the most appropriate PEEP for each patient. Thereby, due to the importance of transpulmonary pressure in lung injury, an obvious approach would be to use transpulmonary pressure to set the PEEP, with the use of esophageal pressure as a surrogate for pleural pressure [57].
However, in this case is difficult obtain an adequate interpretation of absolute esophageal pressure for various reasons, including cardiac artifacts, the uneven distribution of pleural pressure, and esophageal distortion and contraction (especially in supine patients) [75]. However, a pilot study in ARDS patients evaluated this approach [76], setting the PEEP to achieve an end-expiratory transpulmonary pressure of 0 to 10 cm of water, while limiting end-inspiratory transpulmonary pressure to 25 cm of water. Thus, they found an improved oxygenation and a trend toward lower 28-day mortality [76]. However, it is necessary a larger clinical trial that shows significant clinical results, before this approach can be recommended in clinical practice. Finally, the recruitment maneuvers, that are based on the ability to achieve inflate atelectatic regions minimizing the ventilation heterogeneity, should theoretically reduce the lung injury [77]. Although some clinical trials that used such maneuvers have been included in a meta-analysis [74], and have been implemented as a protective strategy increasing the number of lungs retrieved from heart-beating donors [78]. The role of recruitment maneuvers in clinical practice remains uncertain because of questions about its effect on outcomes and concerns regarding complications such as hemodynamic compromise or pneumothorax [3].
4.3. High-Frequency Oscillatory Ventilation
High-frequency oscillatory ventilation [HFOV] is a strategy characterized by use very small tidal volumes, applied at high frequencies (up to 15 per second) [57], which should theoretically be ideal to minimizing ventilator-induced lung injury [79]. A meta-analysis which included eight clinical trials involving a total of 419 adults with ARDS, was found that HFOV-treated patients had significantly lower mortality than patients treated with conventional ventilation, which suggested that HFOV might improve survival and is unlikely to cause damage [80]. However, due to two recent large multicenter trials in ARDS patients did not show improved outcomes with HFOV [38, 39], this ventilatory strategy cannot be recommended as first-line therapy in such patients [81].
CONCLUSION
The acute respiratory distress syndrome (ARDS) persists as an important health problem in intensive care units, responsible of a significant co-morbidity and mortality [1]. The pathophysiology of VILI has initial physical mechanisms, which subsequently through the mechanotransduction lead to a molecular damage response. The physical mechanisms of injury include two main phenomena which may be overlapping, the over-distension in case of an unnecessarily high PEEP (volutrauma) and atelectrauma caused by the cyclic alveolar collapse and reopening in patients with excessive VT reduction (Fig. 1).
The mechanotransduction signals, lead to a series of events including development of inflammation, necrosis, apoptosis and remodeling of the extracellular matrix. Several ventilatory strategies have been evaluated to prevent lung injury, but so far only the reduction of VT to prevent volutrauma and an adequate PEEP to prevent atelectrauma have mostly been validated for use in routine clinical practice. Other potential ventilatory strategies need to be evaluated by larger clinical studies before extending its use in clinical practice.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Author’s Role: All authors helped to write the manuscript and have seen and approved the final version.

