All published articles of this journal are available on ScienceDirect.

Metabolic Alkalosis in Adults with Stable Cystic Fibrosis
Abstract
Background:
The frequency of metabolic alkalosis among adults with stable severe CF-lung disease is unknown.
Methods:
Retrospective chart review.
Results:
Fourteen CF and 6 COPD (controls) patients were included. FEV1 was similar between the two groups. PaO2 was significantly higher in the COPD (mean ± 2 SD is 72.0 ± 6.8 mmHg,) than in the CF group (56.1 ± 4.1 mmHg). The frequency of metabolic alkalosis in CF patients (12/14, 86%) was significantly greater (p=0.04) than in the COPD group (2/6, 33%). Mixed respiratory acidosis and metabolic alkalosis was evident in 4 CF and 1 COPD patients. Primary metabolic alkalosis was observed in 8 CF and none of the COPD patients. One COPD patient had respiratory and metabolic alkalosis.
Conclusions:
Metabolic alkalosis is more frequent in stable patients with CF lung disease than in COPD patients. This might be due to defective CFTR function with abnormal electrolyte transport within the kidney and/ or gastrointestinal tract.
BACKGROUND
Cystic fibrosis (CF) is a multi-system disease that is caused by mutations in a 230 kb gene on chromosome 7 encoding 1480 aminoacid polypeptide, named cystic fibrosis transmembrane regulator (CFTR) [1]. The mutation in CFTR leads to a defect in ion transport across the respiratory epithelium, including decreased cyclic adenosine monophosphate (cAMP)-dependent chloride transport. Furthermore, sodium absorption is enhanced, possibly due to the effect of epithelial sodium channels (ENaC) on CFTR-mediated ion transfer [2].
Holland et al [3] reported that during respiratory exacerbations, patients with end-stage CF were significantly more frequently alkalotic than were those with chronic obstructive pulmonary disease (COPD). A mixed respiratory acidosis and metabolic alkalosis was evident in 71% of CF patients and 22% of COPD patients (p < 0.01).
In our study, we report the frequency of metabolic alkalosis in stable CF patients with severe pulmonary involvement, compared to patients with stable COPD with comparable airway obstruction. We hypothesized that the frequency of metabolic alkalosis in stable CF patients will be greater than the frequency of metabolic alkalosis in stable COPD patients.
METHODS
After obtaining ethics approval, a retrospective chart and database review was performed on clinically stable CF patients (Toronto CF Database) and COPD patients (Chest Database) at St. Michael’s Hospital, Toronto, Canada. Data collection took place between January 2004 and June 2009. A registered respiratory therapist and a medical resident confirmed the clinical stability of the CF and COPD patients.
Participants in both groups had arterial blood gas (ABG) and serum samples collected for electrolytes and albumin. For the evaluation of the acid-base disturbances, ABG results were encountered. Medications and comorbidities were recorded from the chart review. We also collected data of serum hemoglobin (Hb) and hematocrit (Hct) from complete blood counts to assess for evidence of hypovolemia, where available. Pulmonary function testing was carried out as part of the patients’ usual care at the time of clinical stability.
All participants had already consented to be included in the Database. Inclusion criteria to both groups were age ≥ 18 years, clinical stability and/or initiation of long-term home oxygen therapy, and forced expiratory volume in 1 second (FEV1) ≤ 40% of predicted. Participants were included in the study only if they had FEV1 measurement and blood tests collected within 2 months of the ABG.
Exclusion criteria were pregnancy and patients who have undergone lung transplantation. We did not exclude patients with liver disease, CF-related diabetes (CFRD), or patients with sputum culture positive for Burkholderia cepacia complex.
To identify participants with metabolic alkalosis, we used the Stinebaugh-Austin diagram [4]. The acid-base data was plotted on this diagram to ensure that the metabolic alkalosis is a primary disorder, beyond any renal response to a possible respiratory acidosis. During this analysis, we used the venous HCO3 instead of the arterial HCO3 level. The final results were extracted from this diagram as the study primary outcome.
The statistical examination of differences between groups was done using unpaired t-test for parametric data and the X2 test for nonparametric data, using a Type I error rate of 0.05 and power of 0.90. Means, standard deviations (SD), and further statistical analyses were calculated using Statistical Package for the Social Sciences (SPSS) 15.0® program.
RESULTS
We identified 19 eligible CF patients during the study period. After excluding patients with incomplete blood work results, 20 patients were involved in the study, 14 in the CF group and 6 in the COPD group. Table 1describes subjects’ demographics, FEV1, ABG results, and serum electrolytes.
Baseline Participants’ Variables*
| Variables | CF Group (n = 14) | COPD Group (n = 6) | p-Value |
|---|---|---|---|
| Female (%) | 5 (36%) | 3 (50%) | NS |
| Age (years) | 26.0 (5.8) | 63.17 (7.62) | < 0.001 |
| FEV1 (Liters) | 0.85 (0.2) | 0.70 (0.16) | NS |
| FEV1 (% Predicted) | 25.0 (4.02) | 31.0 (7.16) | NS |
| pH | 7.40 (0.01) | 7.38 (0.03) | NS |
| PaCO2 (mmHg) | 48.07 (5.47) | 47.83 (9.83) | NS |
| PaO2 (mmHg) | 56.07 (4.08) | 71.67 (6.8) | 0.04 |
| Arterial HCO3 (mmol/L) | 29.0 (2.25) | 28.67 (4.13) | NS |
| BE | 4.08 (2.1) | 2.15 (2.54) | 0.09 |
| Sodium (mmol/L) | 136.78 (1.72) | 138.83 (1.94) | 0.05 |
| Potassium (mmol/L) | 4.24 (0.41) | 4.28 (0.3) | NS |
| Chloride (mmol/L) | 97.36 (3.67) | 101.17 (5.98) | 0.004 |
| Venous HCO3 (mmol/L) | 30.57 (3.0) | 28.33 (6.8) | NS |
| Serum Albumin (g/L) | 31.64 (5.93) | 34.33 (4.68) | NS |
* Measurements except for gender are reported means and standard deviations in parentheses.
Mean age was statistically significantly lower in the CF group, compared to the COPD group. Mean FEV1 was not significantly different between the two groups. All blood gas results were compensated with a pH within the normal limits. In the CF group, a primary metabolic alkalosis was observed in 8 participants (57%). Mixed respiratory acidosis and metabolic alkalosis was evident in 28% (4/14) of subjects. The remaining 2 participants had primary respiratory acidosis. In the comparison COPD group, only 33% (2/6) had metabolic alkalosis. Both of these subjects also had respiratory alkalosis. The remaining subjects had either normal values or other acid-base disturbance. The acid-base interpretations are plotted on Stinebaugh-Austin diagram (Fig. 1).
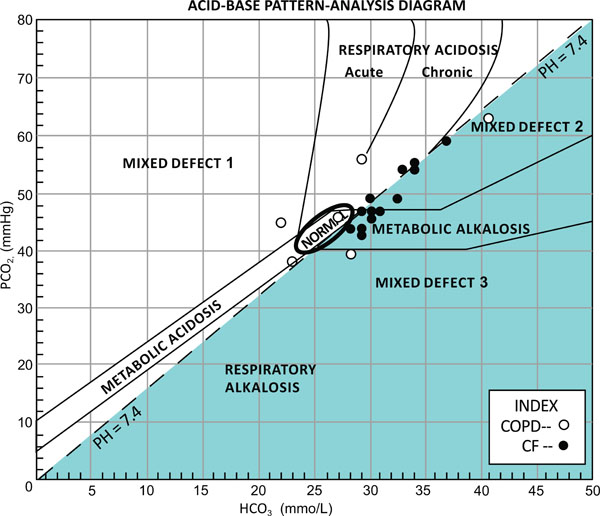
The acid-base findings plotted on Stinebaugh-Austin diagram [4]. Where mixed defects are as follows: 1, metabolic and respiratory acidosis; 2, respiratory acidosis and metabolic alkalosis; and 3, metabolic and respiratory alkalosis.
Mean partial pressure of carbon dioxide (PaCO2) was not significantly different between the CF group and the COPD group. Mean partial pressure of arterial oxygen (PaO2) was significantly greater in the COPD group compared with the CF group (p = 0.04).
Blood biochemical measures revealed statistically significantly lower sodium and chloride levels in the CF patients, but not significantly different albumin level.
Eleven participants had data available for Hb and Hct, 8 from the CF group and 3 from the control group. The mean ± 2 SD for the CF patients was 14.52 ± 1.22 g/dL for Hb, and 44 ± 4% Hct. Values for the Hb and Hct in the control group were 14.2 ± 1.8 g/dL and 44 ± 9%, respectively. Both the Hb and Hct measurements were not statistically different between the 2 groups.
None of the subjects was on diuretics or alkalinizing agents at the time of the testing. One patient in the control group was on steroid therapy. There was no recent history of vomiting or diarrhea in any of the subjects.
DISCUSSION
In this study, we found that metabolic alkalosis is more frequent in adult patients with stable, severe CF lung disease compared with patients with stable COPD of comparable disease severity. Our study confirms previous observations of increased frequency of metabolic alkalosis in CF patients [5-7].
Holland and colleagues [3] reported increased frequency of metabolic alkalosis in CF patients presenting with acute respiratory exacerbations. There is also small number of case reports of CF patients initially presenting with metabolic alkalosis [8-10]. Simple metabolic alkalosis was demonstrated in 16 and mixed metabolic alkalosis and respiratory acidosis in 9 patients [11], though it is unclear whether the patients were stable or acutely ill. Disease stability was not defined in this case series. Our findings, however, suggest that the underlying mechanism of metabolic alkalosis is part of the chronic disease process and not the acute exacerbation per se. Moreover, our study confirms that this predisposition is present in chronic stable disease, rather than only during acute exacerbations.
In our opinion, there are two other plausible hypotheses for the development of metabolic alkalosis in CF patients with a stable disease:
- CF patients may develop contraction alkalosis. Patients with CF are at higher risk of developing body fluid volume contraction and dehydration. This is due to the unique salt (low chloride, high HCO3) losing state [12]. In infants with CF, hyponatremic, hypochloremic dehydration with metabolic alkalosis has been reported. These cases are usually difficult to correct and treatment needs modified oral rehydration solution [13-15]. Patients with CF are also at risk of increased gastrointestinal and insensible fluid losses [16]. In addition, 5-17 % of CF patients are at risk of developing CFRD due to pancreatic endocinopathy [17-20]. Polyuria secondary to CFRD can worsen hypovolemia. Eventually, contraction alkalosis develops. In our study, however, we did not observe evidence of dehydration and so believe that this is not an important mechanism for metabolic alkalosis in stable CF patients.
- Our second hypothesis is that the increased frequency of metabolic alkalosis may be due to an underlying renal process involving CFTR. This receptor is described to be involved in the epithelial cells lining the lung, pancreas, liver, intestines, sweat duct, and the epididymis [21-23]. CFTR regulates and participates in the transport of electrolytes across epithelial-cell membranes and probably across intracellular membranes as well [2,24,25].
CFTR has multiple functions involving fluid balance across epithelial cells. It acts as a chloride channel activated by cAMP [2,26] and may stimulate other chloride channels [27] and inhibit sodium absorption by ENaC [28]. Abnormal ENaC functioning at the level of the kidney in CF patients may precipitate electrolyte disturbances and metabolic alkalosis. CFTR is also related to the HCO3 transport [29,30]. Mucus release in the small intestine is enhanced by HCO3 [31].
We identified limitations in our study design. Given the retrospective nature of the study our recruitment was limited by missing data, especially in the control group. However, given the large difference found between the groups, we believe the conclusions remain valid. We were also limited by missing data in assessing fluid status, and therefore our discussion of mechanisms remains speculative.
We conclude from our data that metabolic alkalosis is more frequent in stable patients with severe CF lung disease than in COPD patients with comparable disease severity, which may be due to CFTR inhibition of ENaC in the kidney. Furthermore, this can be also due to the association between CFTR and bicarbonate transport in the kidney. Larger prospective studies are required to confirm these observations and to clarify the underlying mechanism for this acid-base disturbance.
AUTHORS' CONTRIBUTIONS
Author FA collected the data and drafted the manuscript. Authors FA, MEF, and DET revised and approved the final manuscript.
ACKNOWLEDGEMENTS
We would like to thank Mr. Brent West from the department of respiratory therapy and Mrs. Myra Slutsky for their valuable contribution in allocating participants. Mr. West provided technical support as well. We would also like to thank Dr. Kamel Kamel from the nephrology department for his valuable input and feedback during the preparation of this manuscript.
CONFLICT OF INTEREST STATEMENT
Authors FA and DET have no conflicts of interest to disclose. Financial Support for MEF: Nelson Arthur Hyland Foundation

