All published articles of this journal are available on ScienceDirect.

Hypercapnia Accelerates Wound Healing in Endothelial Cell Monolayers Exposed to Hypoxia
Authors Info & Affiliations
Abstract
Introduction:
While tissue hypoxia is known to play a critical role in the process of vascular injury and repair, the effect of hypercapnia on this process remains uncertain. We investigated whether hypercapnia might influence endothelial cell wound healing under the influence of hypoxia.
Materials and Methodology:
Monolayers of human umbilical venous endothelial cells (HUVECs) were scratch-wounded and incubated under different levels of O2, CO2, and pH in the environment.
Results:
Inhibition of wound healing was observed in the HUVEC monolayers under the hypoxic condition as compared to the normoxic condition. Both hypercapnic acidosis and buffered hypercapnia, but not normocapnic acidosis improved the rate of wound healing under the influence of hypoxia. The beneficial effect of hypercapnia was associated with stimulation of cell proliferation, without effects on cell adhesion, migration or apoptosis. On the other hand, the stimulatory effect of hypercapnia on wound healing and cell proliferation was not noted under normoxic conditions.
Conclusion:
These results suggest that hypercapnia, rather than acidosis per se, accelerated the wound healing in HUVEC monolayers cultured under hypoxic conditions. The effect of hypercapnia on wound healing was due, at least in part, to the stimulation of cell proliferation by hypercapnia.
1. INTRODUCTION
Endothelial cells serve as a barrier between the intravascular compartment and the surrounding tissues. As such, they are directly exposed to various stresses, including hypoxia, ischemia, hyperthermia, and insufficient nutrient supply. If the offending stress is sufficiently severe as to cause endothelial injury, wound repair processes are immediately initiated to restore this barrier function. Wound repair is a complex process that includes cell adhesion, migration, and proliferation, and is influenced by numerous extrinsic and intrinsic factors. Hypoxia is one of the most critical parameters in the regulation of wound healing [1,2] and has been shown to inhibit endothelial wound repair as a result of decreased migration and proliferation [3-6].
Hypoxia is frequently associated with hypercapnia in a range of clinical conditions, such as chronic obstructive pulmonary disease, neuromuscular diseases, and sleep apnea syndrome [7,8]. Furthermore, advance in our understanding of the role of excessive tidal volume has prompted clinicians to use ventilation modes that would result in hypercapnia [9-11]. Consequently, hypercapnia has become increasingly prevalent in the critically ill patient. In patients with acute lung injury/acute respiratory distress syndrome (ALI/ARDS), therapeutic hypoventilation has been reported to be associated with improved patient outcomes, by reducing mechanical damage and lung inflammation [9-11]. The protective action of hypercapnia and/or hypercapnic acidosis is thought to be due, at least in part, to its anti-inflammatory effects [8-10] including attenuation of neutrophil functions [12-14], reduction of free radical generation [15], decreased oxidant-induced tissue damage [16], suppression of nuclear factor kappa B (NFκB) activation [17,18] and reduction in the levels of proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1 [19] and IL-8 [13]. However, the pathophysiological effects of hypercapnia and/or hypercapnic acidosis on wound healing are incompletely understood.
Since endothelial cells are the initial cells within the vascular wall exposed to changes in the ambient O2 and CO2 concentrations in the blood, we explored the effects of hypoxia with or without hypercapnia on endothelial wound healing in vitro. We report here that the healing process of scratched endothelial monolayers was suppressed under hypoxia, whereas the presence of hypercapnia in addition to hypoxia promotes wound healing by stimulating cell proliferation. Our findings suggest that hypercapnia supports the maintenance of endothelial integrity and homeostasis that have been compromised under hypoxic conditions.
2. MATERIALS AND METHODOLOGY
2.1. Cell Culture
Human umbilical venous endothelial cells (HUVECs) were purchased from Lonza (Walkersville, MD) and cultivated in EGM-2 BulletKit supplemented with 2% FBS (growth medium) at 37°C in a humidified incubator saturated with a gas mixture containing 21% O2 and 5% CO2 in N2 (normocapnic normoxia) for up to 5 passages. The initial pH of the EGM-2 medium saturated with 21% O2 and 5% CO2 at 37°C was 7.3; the medium was used after the pH was adjusted to 7.4 by the addition of 0.1N NaOH solution. The exposures of the cells to hypoxia and/or hypercapnia were carried out by incubation of the HUVECs in humidified atmospheres containing 5% CO2 and 1% O2 (normocapnic hypoxia), 10% CO2 and 1% O2 (hypercapnic hypoxia), and 10% CO2 and 21% O2 (hypercapnic normoxia), with the balance composed of N2 in each case, at 37oC. The pH of the medium saturated with 10% CO2 and either 1% O2 or 21% O2 was 7.1, modeling acute hypercapnia. In some experiments modeling chronic hypercapnia, 0.1N NaOH solution was added to the medium saturated with 10% CO2 and either 1% O2 or 21% O2, to obtain a final pH of 7.4. In some additional experiments modeling metabolic acidosis, 0.5N HCl solution was added to the medium saturated with 5% CO2 and either 1% O2 or 21% O2, to obtain a final pH of 7.1. The medium was presaturated by incubation in the appropriate gas mixture overnight, and then added to the cell cultures, as indicated, in each experiment.
2.2. In Vitro Wound Healing Assay
HUVECs were grown to confluence in 8-chamber tissue culture dishes. Thereafter, the monolayers were wounded in a straight line using a sterile 200 μL tip and washed twice with phosphate-buffered saline (PBS). The monolayers were quickly added to the presaturated medium and the incubation was continued in the appropriate gas mixture until 48 hours post injury. The progress of wound healing was monitored with a phase-contrast Olympus BX60 microscope (Olympus Optical Co., Ltd., Tokyo, Japan) equipped with a digital camera. Each wound was photographed at specified time-points, and the area of the wound was measured with an image analysis software (Win Roof Version 3.5; Mitani Corporation, Fukui, Japan) on a Microsoft XP computer. The dishes were returned to the incubator between the measurements. Four measurements were taken from four fields of each well in at least three independent experiments.
2.3. Adhesion Assay
HUVECs at subconfluence were trypsinized, resuspended in the medium presaturated with each of the appropriate gas mixtures, and seeded on to 96-well dishes. After 1 hour of incubation in the same gas mixture condition, non-attached cells were removed by a gentle wash with PBS. Then, the attached cells were lysed in 100 µl of PBS containing 0.2% Triton-X, in which the total protein concentration was measured by the Protein DC assay (Bio-rad Laboratories, Tokyo, Japan) according to the manufacturer’s instructions. The adhesion was quantified by dividing the protein concentration in the attached cell lysate by the protein concentration in the total cell lysate.
2.4. Cell Migration Assay
Cell migration was examined by the Boyden chamber technique using a 48-well chamber (Neuroprobe, Bethesda, MD). Polycarbonate membranes with 8-µm pores (Neuroprobe) were coated with 1% collagen. Vascular endothelial growth factor (VEGF; 500 ng/ml) was used in the bottom wells as an attractant. HUVECs at subconfluence were trypsinized, resuspended in in the presaturated medium, and plated on to each of the top wells above the filter. The chambers were then incubated for 6 hours under each gas mixture condition. After incubation, cells on the top of the filter were removed by scraping. The filter was then stained with a Diff-Quick stain. Endothelial cell migration activity was quantified as the number of migrated cells on the lower surface of the filter in 10 high-power fields using a light microscope at x 1000 magnification.
2.5. Cell Proliferation Assay
Proliferating HUVECs were identified by immunocytochemistry for the detection of bromodeoxyuridine (BrdU) incorporation into the cellular DNA. HUVECs at 50% confluence were incubated at different O2, CO2, and pH conditions for different periods of time. Cells were loaded with 30 µM BrdU for the final 1 hour and then fixed for immunostaining to detect cell proliferation as described below. Cell proliferation was quantified by counting BrdU-incorporated cells under a light microscope at x400 magnification.
2.6. Immunocytochemistry
HUVECs were fixed with 3% paraformaldehyde and permeabilized with 0.5% Triton X-100. For anti-BrdU staining, cells were exposed to 2N HCl for 20 minutes to denature DNA for easy access of the antibody to antigen. Nonspecific binding was blocked with a blocking solution containing 3% BSA, followed by incubation with mouse monoclonal anti-BrdU antibody (Merck Milipore Co., Billerica, MA) or rabbit polyclonal anti-active caspase-3 antibody (Cell signaling Technology Japan, Tokyo, Japan). The primary antibodies were reacted with the secondary antibodies conjugated with horseradish peroxidase polymer (EnVision+ kit; Dako Japan, Tokyo, Japan). The immunoreactants were detected with a 3,3’-diaminobenzidine solution.
2.7. TUNEL
TdT-mediated dUTP nick-end labeling (TUNEL) was performed using the Takara In Situ Apoptosis Detection Kit (Takara Biomedicals, Tokyo, Japan) according to the manufacture’s instructions.
2.8. Statistical Analysis
The statistical analyses were performed using the Excel X software program with the add-in software Statcel 3 (OMS, Tokyo, Japan). Data are expressed as mean ± SEM. Statistical differences were analyzed by analysis of variance (ANOVA), and if the results were significant, the Tukey-Kramer test was used as a multiple comparison post hoc test. P values of <0.05 were considered to indicate statistical significance.
3. RESULTS
3.1. Hypercapnia Accelerates Wound Healing in HUVEC Monolayers Under Hypoxic Conditions
A reduced rate of wound healing was observed in HUVEC monolayers at 24 and 48 hours after injury under normocapnic hypoxia (5% CO2, 1% O2, pH 7.4) as compared to that under the normocapnic normoxic controls (5% CO2, 21% O2, pH 7.4) (Fig. 1). However, hypercapnic acidosis (10% CO2, 1% O2, pH 7.1), modeling acute hypercapnia, improved the rate of wound healing in the hypoxic HUVEC monolayers (Fig. 1B, C). Buffered hypercapnia (10% CO2, 1% O2, pH 7.4), modeling chronic hypercapnia, also improved the rate of wound healing in the hypoxic HUVEC monolayers (Fig. 1B, C). The degree of improvement in the rate of wound healing under buffered hypercapnia was not different from that seen under hypercapnic acidosis. In contrast, normocapnic acidosis (5% CO2, 1% O2, pH 7.1), modeling metabolic acidosis, did not alter the rate of wound healing in hypoxic cultures of HUVEC monolayers (Fig. 1C). Hypercapnia did not stimulate wound healing of the HUVEC monolayers under normoxic conditions (Fig. 1D). These findings suggest that hypercapnia rather than acidosis per se accelerates the wound healing in HUVEC monolayers under hypoxic conditions.
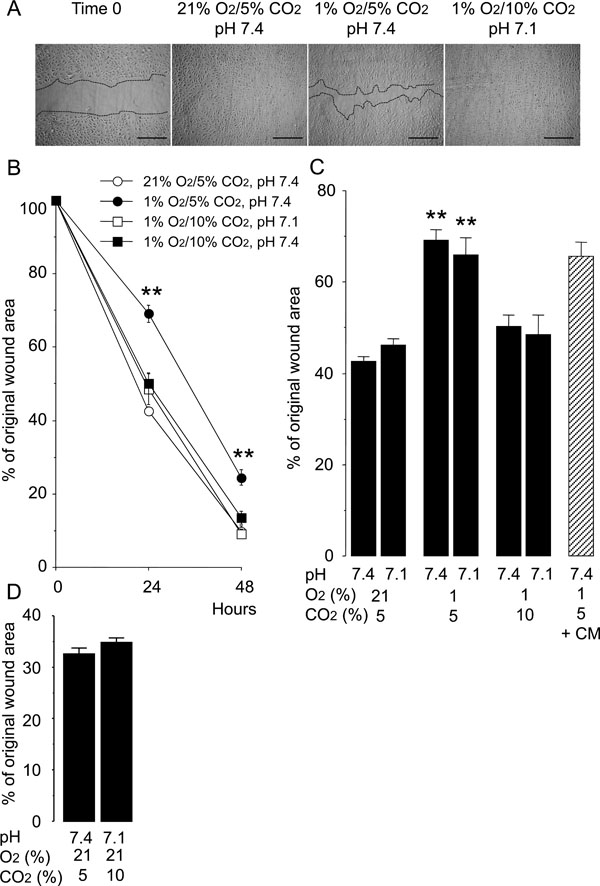
Hypercapnia accelerates wound healing in HUVEC monolayers exposed to hypoxia. Cells were grown to confluence in 8-chamber tissue culture dishes before wounding. (A) Representative photomicrographs of cell monolayers 48 hours after wounding. Broken lines indicate the leading edge of the wound repopulating cells. Scale bars = 200 µm. (B) Time course of changes in the size of the remaining wound. The data points represent the means ± SEM of 4 wells. **P < 0.01 vs cells cultured at 21% O2/5% CO2, pH 7.4; 1% O2/10% CO2, pH 7.1 and 1% O2/10% CO2 buffered to pH 7.4. (C) Effects of hypoxia, hypercapnia and acidosis on wound healing. Wounded cell monolayers were cultured under various conditions with different O2 (21% or 1%), CO2 (5% or 10%), and pH (7.4 or 7.1) levels. The areas of the wounds were measured at 24 hours. Wounded cell monolayers were also cultured at 1% O2/5% CO2, pH 7.4 in conditioned medium (CM) from cell cultures exposed to 1% O2/10% CO2, pH 7.1 for 48 hours. **P < 0.01 as compared to all other conditions. (D) Effect of hypercapnia on wound healing in HUVEC monolayers cultured under normoxia at 24 hours.

Hypercapnia does not stimulate cell attachment (A) or migration (B) of HUVECs exposed to hypoxia. Cells were grown to subconfluence before trypsinization and replated on to 96-well dishes for the 1-hour attachment assay (A) or used in the Boyden blind well chamber migration assay for 6 hours, during which VEGF (500 ng/ml) was used as an attractant (B). Both assays were performed under different cell culture conditions as specified. The bars represent the means ± SEM of 6 wells (A) or 3 wells (B) **P < 0.01 as compared to control cells incubated under 21% O2 and 5% CO2, pH 7.4.
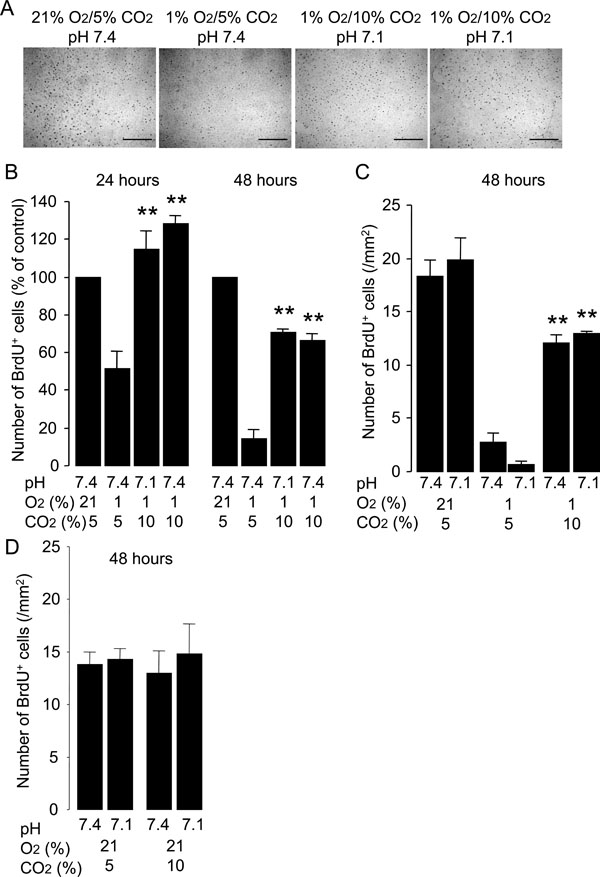
Hypercapnia stimulates proliferation of HUVECs exposed to hypoxia. Cells were grown to 50% confluence and then incubated under various conditions with different O2 (21% or 1%), CO2 (5% or 10%) and pH (7.4 or 7.1) levels for 24 or 48 hours. BrdU was added to the medium 1 hour before paraformaldehyde fixation. (A) Representative photomicrographs of anti-BrdU immunocytochemistry for HUVECs fixed at 24 hours. Distinct dark spots represent cell nuclei with BrdU incorporation. Scale bars = 200 µm. (B) Effect of hypercapnia on cell proliferation. Hypercapnic acidosis and buffered hypercapnia stimulate cell proliferation under hypoxic conditions. Cell proliferation at 24 and 48 hours was quantified as the number of BrdU+ cells/mm2 and data are expressed as percentages of values obtained from the normocapnic normoxic controls (21% O2/5% CO2, pH 7.4) at the corresponding time-points. The bars represent the means ± SEM of 4 wells. **P < 0.01 as compared to cells incubated at 1% O2/5% CO2, pH 7.4. (C) Hypercapnic acidosis but not noromocapnic acidosis stimulates cell proliferation under hypoxic conditions. Data are expressed as the number of BrdU+ cells/mm2. The bars represent the means ± SEM of 4 wells. **P < 0.01 as compared to cells incubated under conditions of 1% O2/5% CO2 with pH 7.4 or pH 7.1. (D) Hypercapnia did not stimulate cell proliferation under noromoxic conditions. Data are expressed as the number of BrdU+ cells/mm2. The bars represent the means ± SEM of 4 wells.
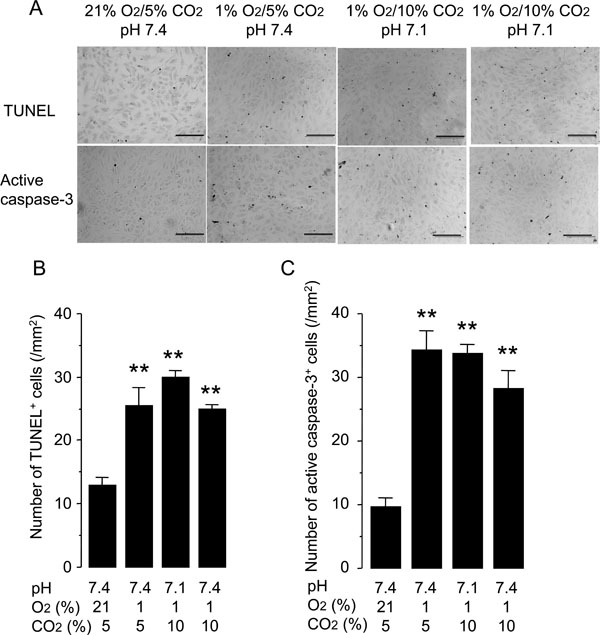
Hypercapnia does not protect against hypoxia-induced apoptosis of HUVECs. Cells were grown to 50% confluence and incubated under conditions with different O2 (21% or 1%), CO2 (5% or 10%), and pH (7.4 or 7.1) levels for 48 hours. Apoptosis was assessed by TUNEL staining and anti-active-caspase-3 immunocytochemistry. (A) Representative photomicrographs of TUNEL and anti-active-caspase- 3 immunocytochemistry. Scale bars = 200 µm. (B) Quantification of TUNEL+ cells. (C) Quantification of anti-active-caspase-3+ cells. The bars represent the means ± SEM of 4 wells. **P < 0.01 as compared to cells incubated at 21% O2/5% CO2, pH 7.4.
The rate of wound healing in HUVEC monolayers cultured under normocapnic hypoxia was not altered by the conditioned medium from HUVEC monolayers cultured under hypercapnic hypoxia for 24 hours (Fig. 1C). This finding suggests that the stimulatory effect of hypercapnia on wound healing was not due to the secretion of a soluble factor from the hypercapnia-exposed HUVECs.
3.2. Hypercapnia Does Not Stimulate Cell Adhesion or Migration of HUVECs Exposed to Hypoxia
Wound healing of cell monolayers involves complex processes, including adhesion, migration, and proliferation. Adhesion and migration of HUVECs was inhibited under normocapnic hypoxic conditions as compared with that in the normocapnic normoxic controls (Fig. 2). Neither hypercapnic acidosis nor buffered hypercapnia altered the rates of adhesion or migration of HUVECs exposed to hypoxia. These results suggest that the acceleration of wound healing by hypercapnia under hypoxic conditions was not due to stimulation of cell adhesion or migration.
3.3. Hypercapnia Stimulates Proliferation of HUVECs Exposed to Hypoxia
The effect of hypercapnia on the proliferation of HUVECs was determined by immunocytochemistry for the detection of BrdU incorporation into the cellular DNA. Normocapnic hypoxia inhibited the proliferation of HUVECs as compared with the proliferative activity observed in the normocapnic normoxic controls (Fig. 3). Both hypercapnic acidosis and buffered hypercapnia stimulated proliferation of HUVECs exposed to hypoxia (Fig. 3B). In contrast, normocapnic acidosis did not alter the rate of cell proliferation observed under hypoxic conditions (Fig. 3C). Neither hypercapnic acidosis nor buffered hypercapnia altered the rate of cell proliferation observed under noromoxic conditions (Fig. 3D). These results suggest that hypercapnia stimulates the proliferation of HUVECs exposed to hypoxia.
3.4. Hypercapnia Does Not Protect Hypoxia-Induced Apoptosis of HUVECs
Another potential mechanism underlying the acceleration of wound healing by hypercapnia was protection of HUVECs from hypoxia-induced apoptosis. Incubation of HUVECs under normocapnic hypoxia for 48 hours increased the rate of apoptosis detected by TUNEL and active caspase-3 immunostaining as compared with that observed in the normocapnic normoxic controls (Fig. 4). Neither hypercapnic acidosis nor buffered hypercapnia altered the rate of apoptosis, suggesting that the effect of hypercapnia on wound healing was not due to modulation of apoptosis.
4. DISCUSSION
The present study demonstrated that hypercapnia, rather than acidosis per se, accelerated wound healing in the HUVEC monolayers cultured under hypoxic conditions. The effect of hypercapnia on wound healing was due, at least in part, to the stimulation of cell proliferation by hypercapnia.
Tissue hypoxia plays a critical role in the process of vascular injury and repair [20]. The pO2 in the alveoli of the lung is 110 mmHg, whereas the median pO2 in the brain, liver, and heart is closer to 24 mmHg (= about 3%). Thus, the hypoxic concentration of 1% O2 that we tested in this study is a tissue hypoxia level that is thought to be often encountered in conditions with reduced pO2 in the arterial blood [21]. The results of the present study regarding the inhibition of endothelial wound healing by hypoxia corroborate those of previous studies [3,4,6]. Hypoxia-induced inhibition of endothelial wound healing has been attributed to inhibition of proliferation [5,22] and migration of the cells [4,6]. However, the role of CO2 in endothelial wound healing has not been examined previously. The present study showed for the first time that hypercapnia supports endothelial wound healing under hypoxic conditions via, at least in part, stimulation of cell proliferation.
Hypercapnia has myriad effects on many physiological processes, and its influence on cell behavior is known to be rather heterogeneous [8,10,23]. Our results are contrary to those of O’Toole et al. [24] who reported that hypercapnia inhibited wound healing in vitro. However, the experimental protocols differed between their studies and ours. O’Toole and colleagues showed that hypercapnia (10% and 15% CO2) under noromoxic conditions retarded wound healing in monolayers of alveolar and bronchial epithelial cells by suppression of cell migration, whereas our data showed that hypercapnia (10% CO2) under hypoxic conditions (1% O2) improved wound healing in endothelial cell monolayers by, at least in part, stimulating cell proliferation. Our results are also inconsistent with those of the studies conducted by Vohwinkel and colleagues [25]. These researchers showed that exposure to hypercapnia (7.5% and 15% CO2) for the longer duration of 6 days than that in our study (48 hours) under normoxic conditions decreased proliferation of A549 alveolar epithelial cells and N12 lung fibroblasts by inducing microRNA-183, which downregulated isocitrate dehydrogenase-2, a key enzyme of the TCA cycle. However, they did not examine the effect of hypercapnia on cell proliferation and wound healing under the influence of hypoxia. In the present study, we observed that hypercapnia stimulated HUVEC proliferation and wound healing only under hypoxic conditions, and not under noromoxic conditions. These conflicting lines of evidence suggest that the cellular responses to hypercapnia may differ according to the types of cells, levels of O2 tension, and duration of hypercapnia.
Hypercapnic hypoxia may result from a variety of diseases, not only those involving the lungs, but also those affecting the neural, muscular and chest wall components of the respiratory system [7]. Hypercapnia has traditionally been recognized as harmful because of its deleterious effects on myocardial, neuromuscular and immune functions. However, recent advances in “protective” ventilator strategies in critically ill patients have revealed the beneficial effects of “therapeutic” hypercapnia, e. g., protection of pulmonary, cardiovascular and neuronal injury [8-10, 17, 26, 27]. Molecular mechanisms underlying tissue protection by hypercapnia remain uncertain, although the involvement of the anti-inflammatory and anti-oxidant effects of hypercapnia has been suggested [8-10]. Furthermore, recent studies have shown that hypercapnia exerts its beneficial effects through the inhibition of NF-κB pathway in an endotoxin-induced inflammation model and a ventilation-induced lung injury model [17, 28]. Future studies are needed to further clarify the pathways mediating hypercapnia-induced tissue protection. Since tissue damage is often complicated by vascular injuries associated with tissue hypoxia [20], we suggest, based on our findings, that hypercapnia may contribute to the promotion of vascular regeneration and tissue repair.
5. CONCLUSION
Hypercapnia appears to exert both beneficial and deleterious effects on wound healing, depending on its level, timing, and duration, as well as the prevailing O2 tension. Our findings suggest that hypercapnia (10% CO2) for up to 48 hours supports the maintenance of endothelial integrity and homeostasis by promoting wound repair that has been compromised under hypoxic conditions.
ABBREVIATIONS
CONFLICT OF INTEREST
The authors declare that they have no competing interests in the subjects of this manuscript.
ACKNOWLEDGEMENTS
KY acknowledges a grant from by Teijin Home Healthcare Limited. KA acknowledges a Grant-in-Aid for Scientific Research from the Ministry of Education, Science in Japan. We are also grateful to Masayuki Shino and Yoshimi Sugimura for their excellent technical assistance.

